

The Finnish genetic heritage in 2022 - from diagnosis to translational research
- PMID: 36285626
- PMCID: PMC9637267
- DOI: 10.1242/dmm.049490
The Finnish genetic heritage in 2022 - from diagnosis to translational research
Abstract
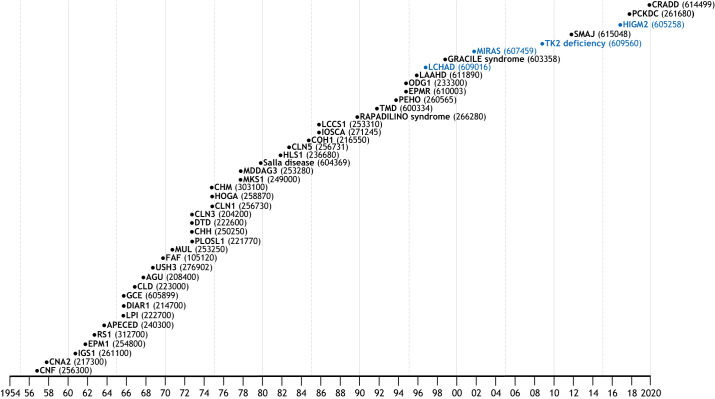
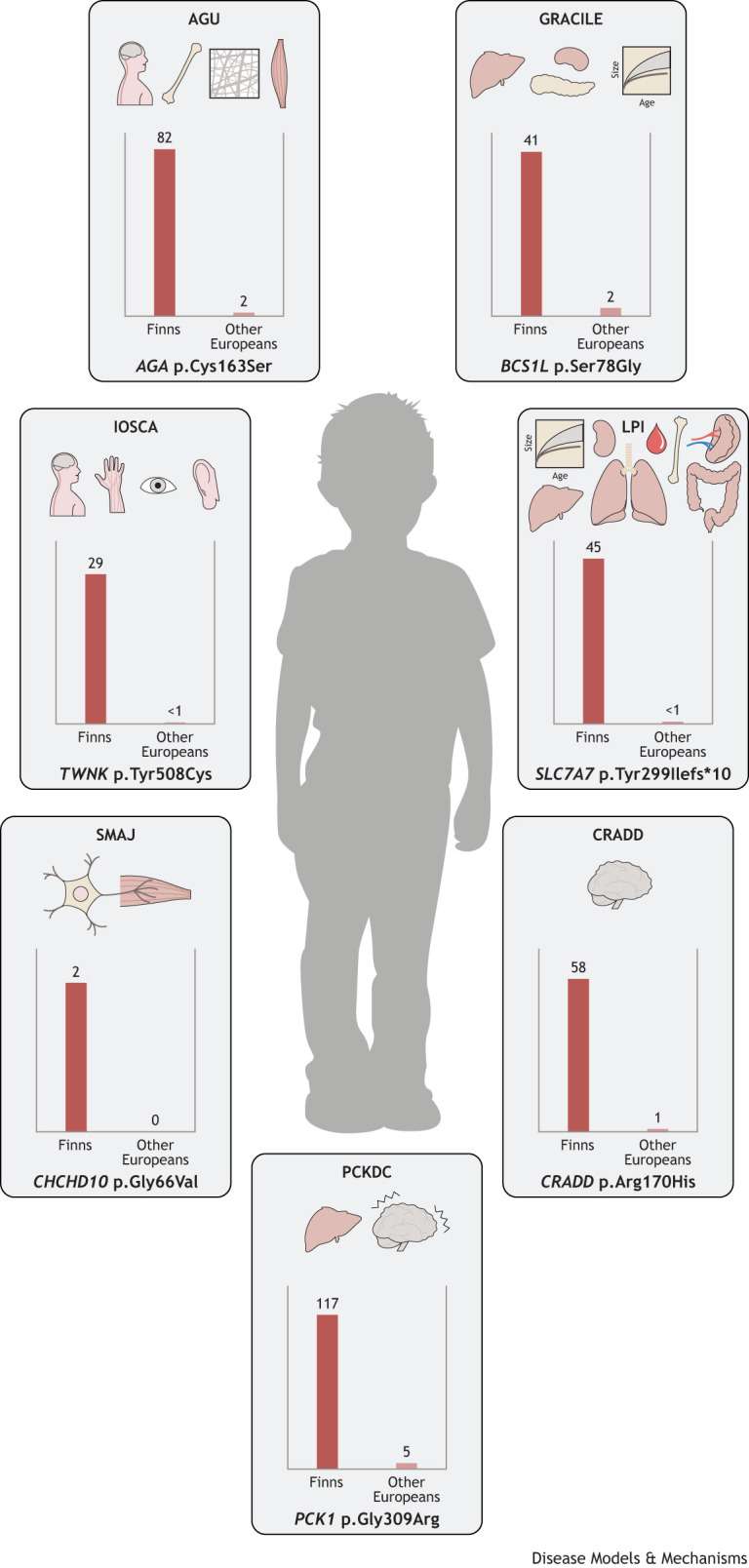
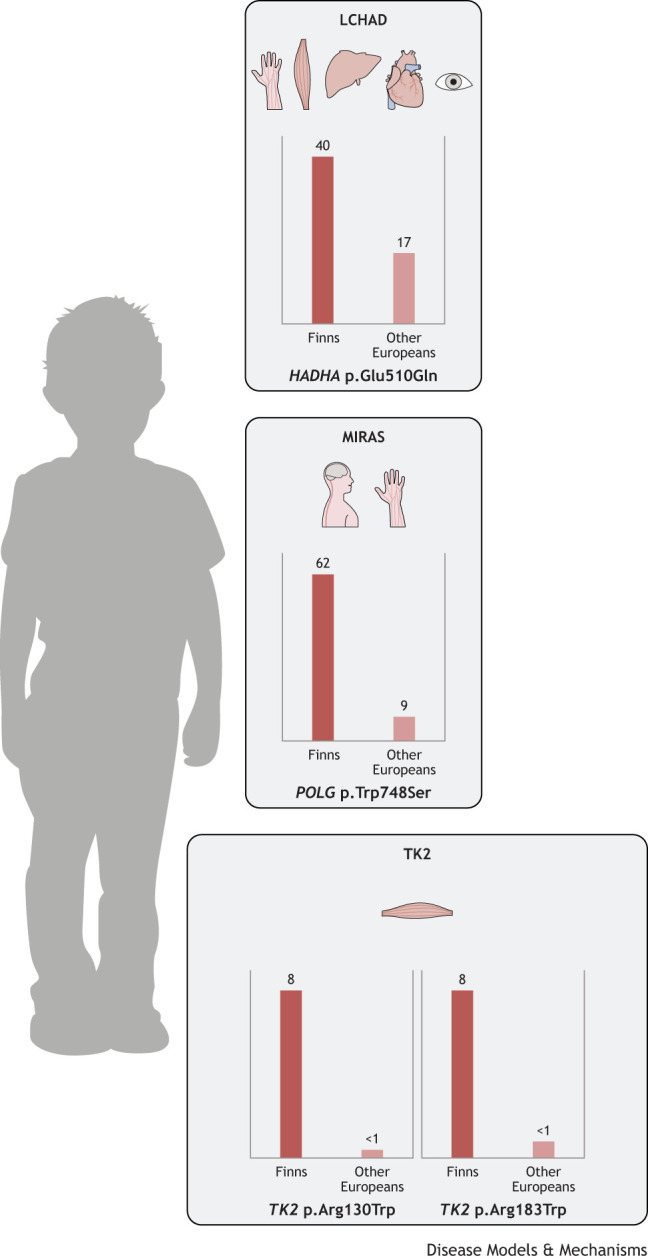
Isolated populations have been valuable for the discovery of rare monogenic diseases and their causative genetic variants. Finnish disease heritage (FDH) is an example of a group of hereditary monogenic disorders caused by single major, usually autosomal-recessive, variants enriched in the population due to several past genetic drift events. Interestingly, distinct subpopulations have remained in Finland and have maintained their unique genetic repertoire. Thus, FDH diseases have persisted, facilitating vigorous research on the underlying molecular mechanisms and development of treatment options. This Review summarizes the current status of FDH, including the most recently discovered FDH disorders, and introduces a set of other recently identified diseases that share common features with the traditional FDH diseases. The Review also discusses a new era for population-based studies, which combine various forms of big data to identify novel genotype-phenotype associations behind more complex conditions, as exemplified here by the FinnGen project. In addition to the pathogenic variants with an unequivocal causative role in the disease phenotype, several risk alleles that correlate with certain phenotypic features have been identified among the Finns, further emphasizing the broad value of studying genetically isolated populations.
Keywords: Big data; FinnGen; Finnish disease heritage; Monogenic disorders; Population isolate; Rare disease.
© 2022. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests R.T. has received research funds from Neurogene Inc. for studies on gene therapy for FDH diseases.
Figures
References
-
- Ahola, S., Mari, A., Pirjo, I., Satu, N., Niina, U., Jana, B., Vidya, V., Lundbom, N., Hakkarainen, A., Muurinen, T.et al. (2016). Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 8, 1234-1247. 10.15252/emmm.201606592 - DOI - PMC - PubMed
-
- Ahvenainen, E. K., Hallman, N. and Hjelt, L. (1956). Nephrotic syndrome in newborn and young infants. Ann. Paediatr. Fenn. 2, 227-241. - PubMed
-
- Ajroud-Driss, S., Faisal, F., Kaouther, A., Irfan, L., Sarah, E. C., Vamsi, K. M., Han, X. D., Siddique, N., Tahmoush, A. J., Heiman-Patterson, T. D.et al. (2015). Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 16, 1-9. 10.1007/s10048-014-0421-1 - DOI - PMC - PubMed
-
- Akman, H. O., Beatriz, D., Luis, C. L., Ángeles, G.-C., Maya, R. V., Lauren, M. T., William, T. D., Eduardo, B., Kurenai, T. and Michio, H. (2008). Thymidine kinase 2 (H126N) Knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum. Mol. Genet. 17, 2433-2440. 10.1093/hmg/ddn143 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
