

Common Germline Risk Variants Impact Somatic Alterations and Clinical Features across Cancers
- PMID: 36286845
- PMCID: PMC9811159
- DOI: 10.1158/0008-5472.CAN-22-1492
Common Germline Risk Variants Impact Somatic Alterations and Clinical Features across Cancers
Abstract
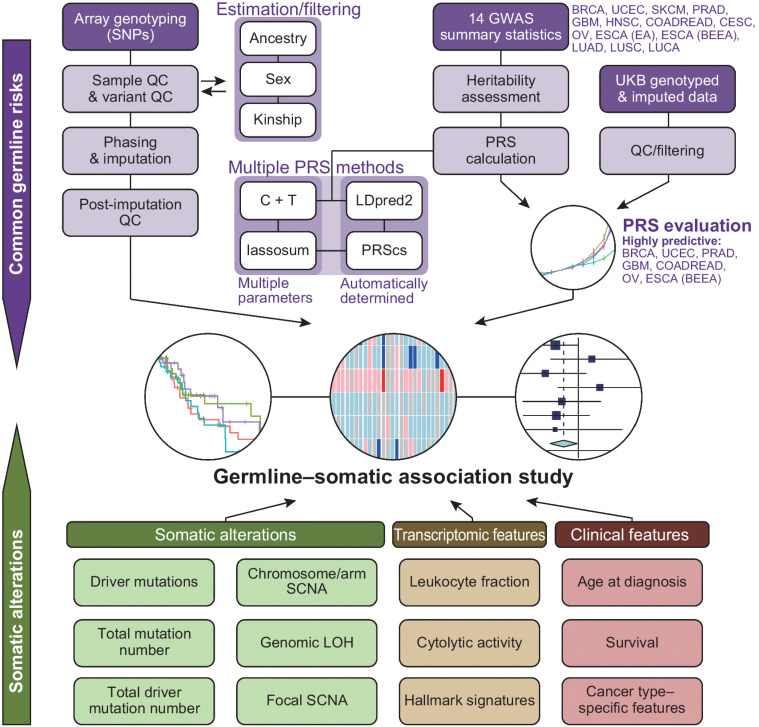
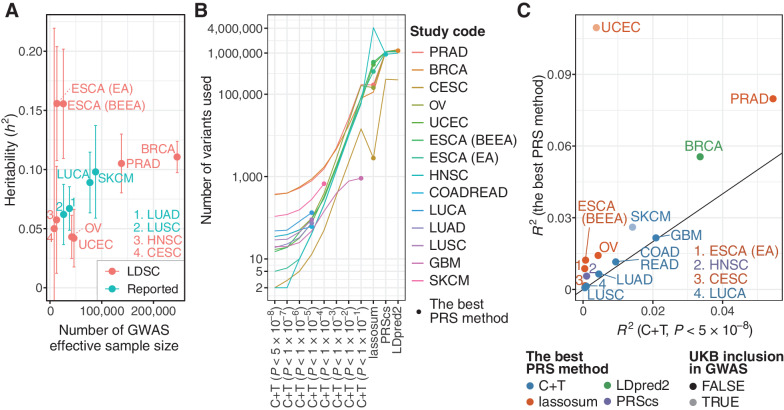
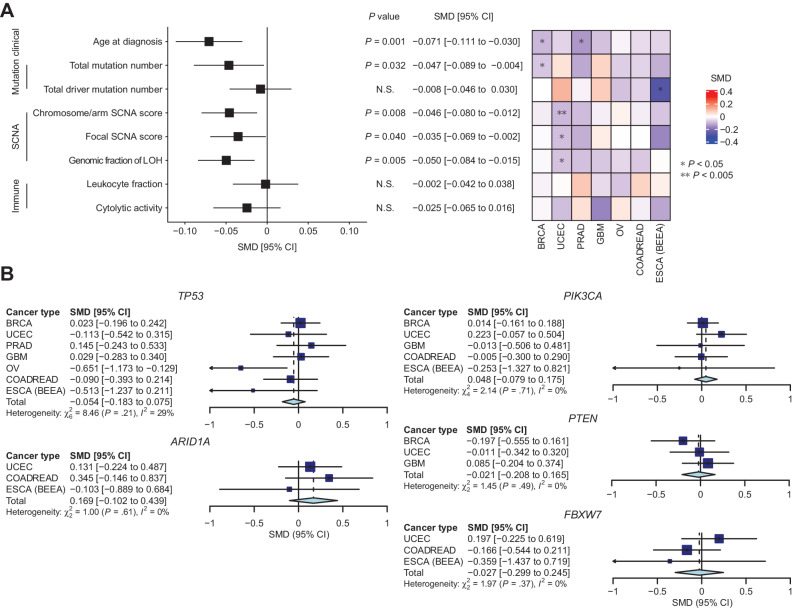
Aggregation of genome-wide common risk variants, such as polygenic risk score (PRS), can measure genetic susceptibility to cancer. A better understanding of how common germline variants associate with somatic alterations and clinical features could facilitate personalized cancer prevention and early detection. We constructed PRSs from 14 genome-wide association studies (median n = 64,905) for 12 cancer types by multiple methods and calibrated them using the UK Biobank resources (n = 335,048). Meta-analyses across cancer types in The Cancer Genome Atlas (n = 7,965) revealed that higher PRS values were associated with earlier cancer onset and lower burden of somatic alterations, including total mutations, chromosome/arm somatic copy-number alterations (SCNA), and focal SCNAs. This contrasts with rare germline pathogenic variants (e.g., BRCA1/2 variants), showing heterogeneous associations with somatic alterations. Our results suggest that common germline cancer risk variants allow early tumor development before the accumulation of many somatic alterations characteristic of later stages of carcinogenesis.
Significance: Meta-analyses across cancers show that common germline risk variants affect not only cancer predisposition but the age of cancer onset and burden of somatic alterations, including total mutations and copy-number alterations.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
