

Multiplexed Target Enrichment Enables Efficient and In-Depth Analysis of Antimicrobial Resistome in Metagenomes
- PMID: 36287061
- PMCID: PMC9769626
- DOI: 10.1128/spectrum.02297-22
Multiplexed Target Enrichment Enables Efficient and In-Depth Analysis of Antimicrobial Resistome in Metagenomes
Abstract
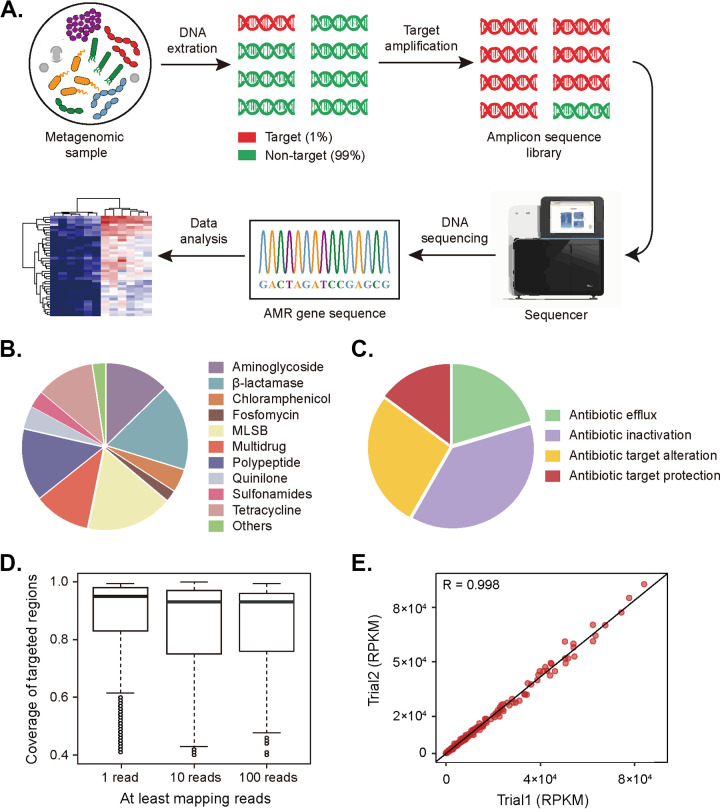
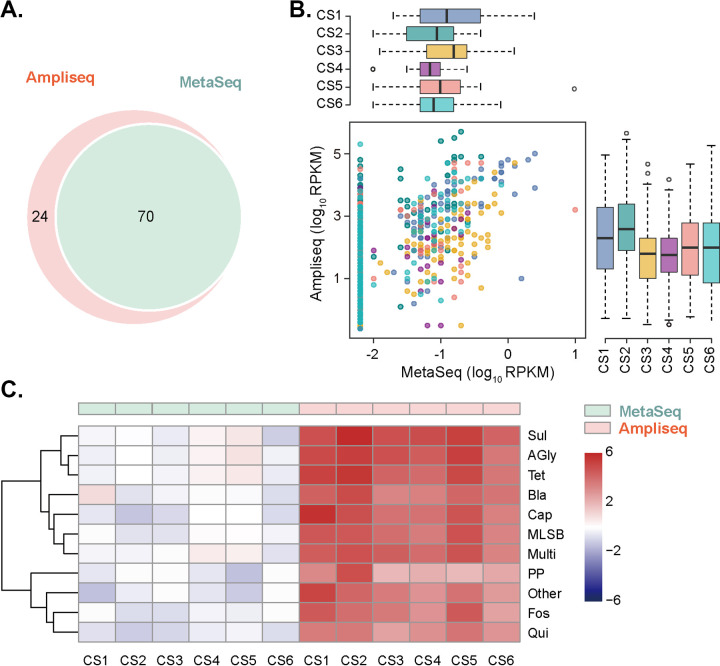
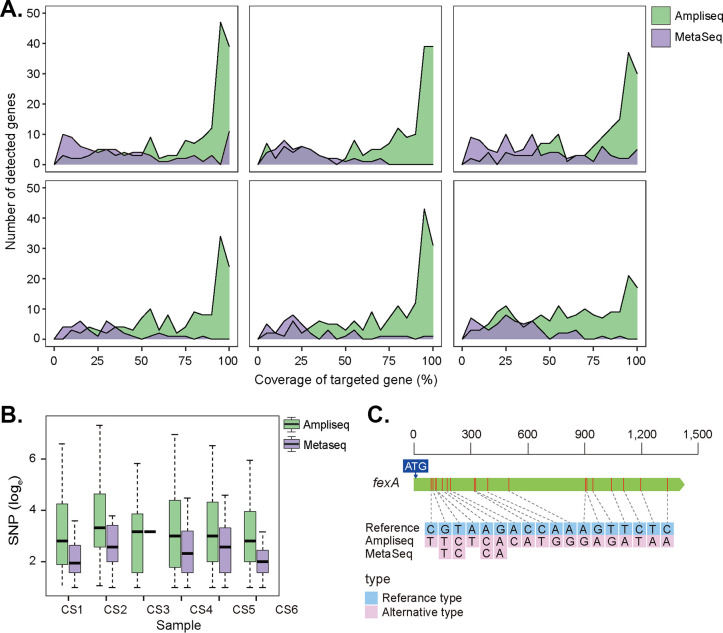
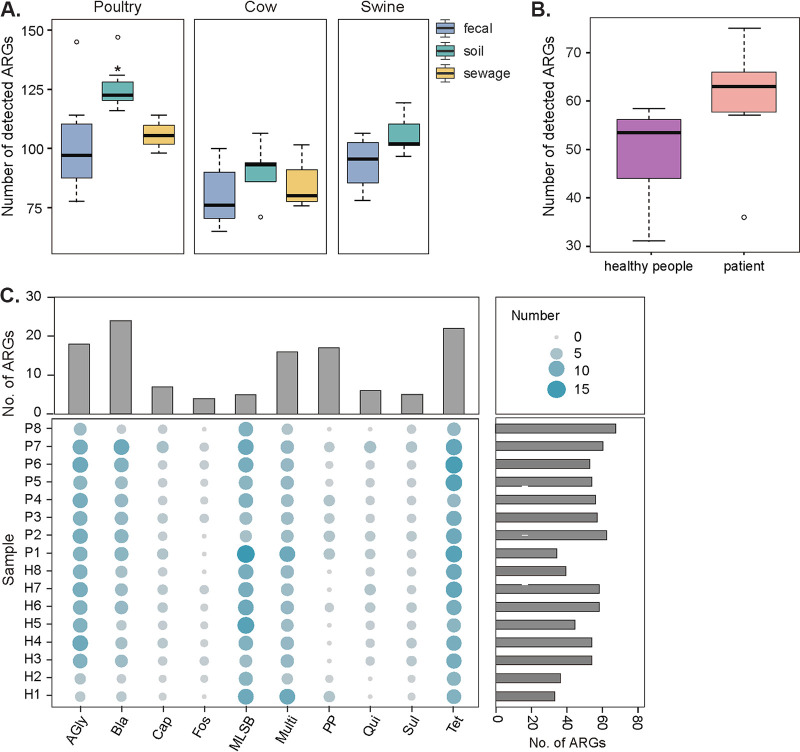
Antibiotic resistance genes (ARGs) pose a serious threat to public health and ecological security in the 21st century. However, the resistome only accounts for a tiny fraction of metagenomic content, which makes it difficult to investigate low-abundance ARGs in various environmental settings. Thus, a highly sensitive, accurate, and comprehensive method is needed to describe ARG profiles in complex metagenomic samples. In this study, we established a high-throughput sequencing method based on targeted amplification, which could simultaneously detect ARGs (n = 251), mobile genetic element genes (n = 8), and metal resistance genes (n = 19) in metagenomes. The performance of amplicon sequencing was compared with traditional metagenomic shotgun sequencing (MetaSeq). A total of 1421 primer pairs were designed, achieving extremely high coverage of target genes. The amplicon sequencing significantly improved the recovery of target ARGs (~9 × 104-fold), with higher sensitivity and diversity, less cost, and computation burden. Furthermore, targeted enrichment allows deep scanning of single nucleotide polymorphisms (SNPs), and elevated SNPs detection was shown in this study. We further performed this approach for 48 environmental samples (37 feces, 20 soils, and 7 sewage) and 16 clinical samples. All samples tested in this study showed high diversity and recovery of targeted genes. Our results demonstrated that the approach could be applied to various metagenomic samples and served as an efficient tool in the surveillance and evolution assessment of ARGs. Access to the resistome using the enrichment method validated in this study enabled the capture of low-abundance resistomes while being less costly and time-consuming, which can greatly advance our understanding of local and global resistome dynamics. IMPORTANCE ARGs, an increasing global threat to human health, can be transferred into health-related microorganisms in the environment by horizontal gene transfer, posing a serious threat to public health. Advancing profiling methods are needed for monitoring and predicting the potential risks of ARGs in metagenomes. Our study described a customized amplicon sequencing assay that could enable a high-throughput, targeted, in-depth analysis of ARGs and detect a low-abundance portion of resistomes. This method could serve as an efficient tool to assess the variation and evolution of specific ARGs in the clinical and natural environment.
Keywords: SNPs; high-throughput sequencing; metagenomics; resistome; targeted amplification.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
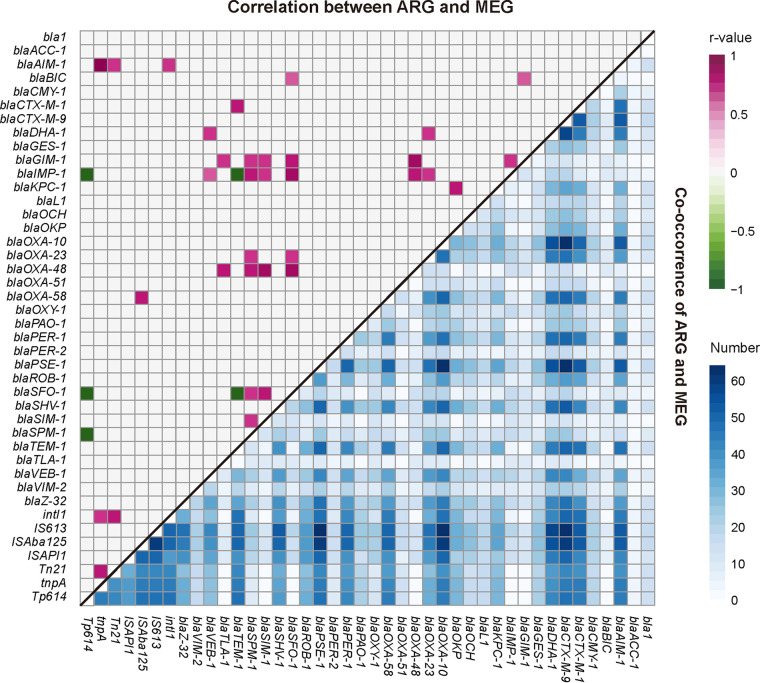
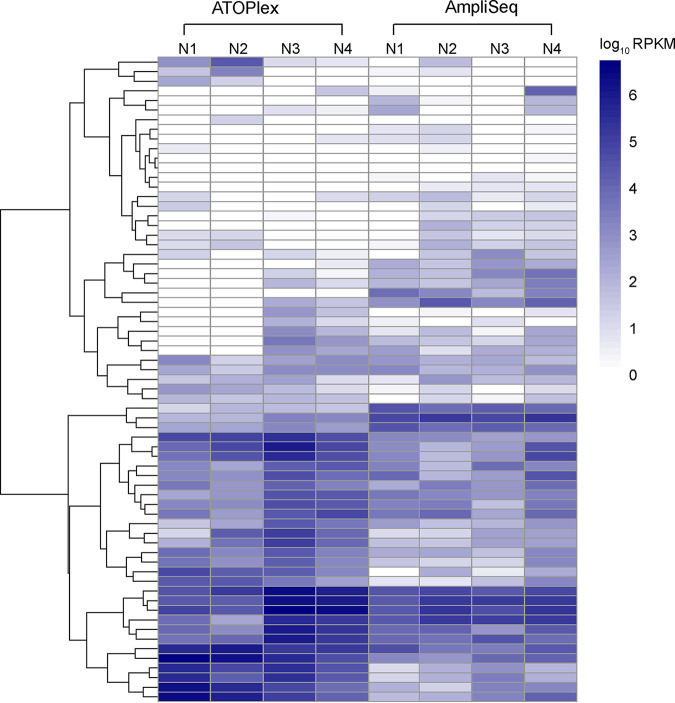
References
-
- WHO. 2019. Ten threats to global health in 2019.
-
- Makkaew P, Kongprajug A, Chyerochana N, Sresung M, Precha N, Mongkolsuk S, Sirikanchana K. 2021. Persisting antibiotic resistance gene pollution and its association with human sewage sources in tropical marine beach waters. Int J Hyg Environ Health 238:113859. doi:10.1016/j.ijheh.2021.113859. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
