

Epigenetic analysis of cell-free DNA by fragmentomic profiling
- PMID: 36288287
- PMCID: PMC9636966
- DOI: 10.1073/pnas.2209852119
Epigenetic analysis of cell-free DNA by fragmentomic profiling
Abstract
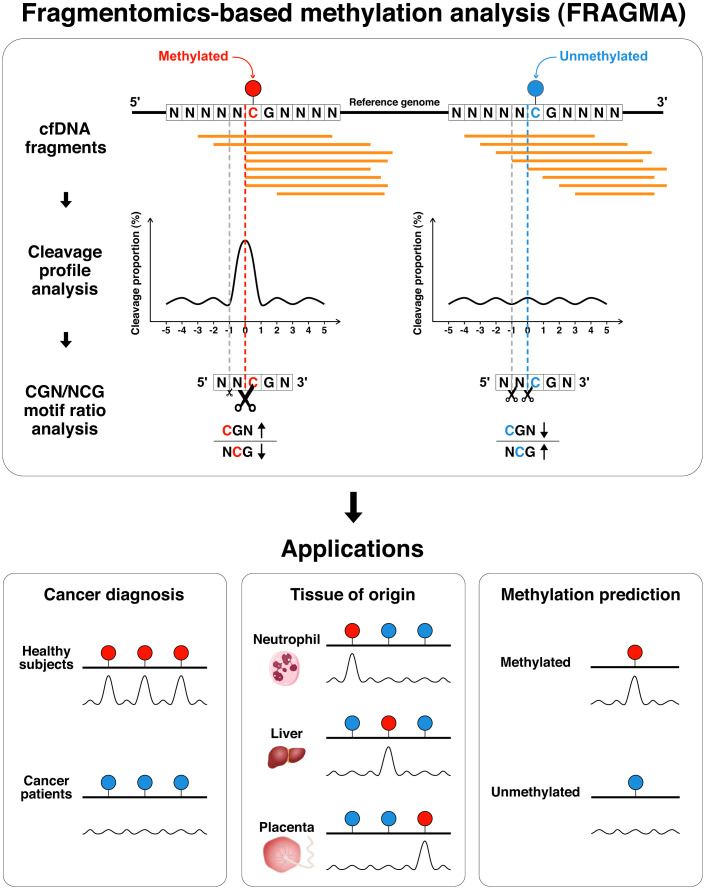
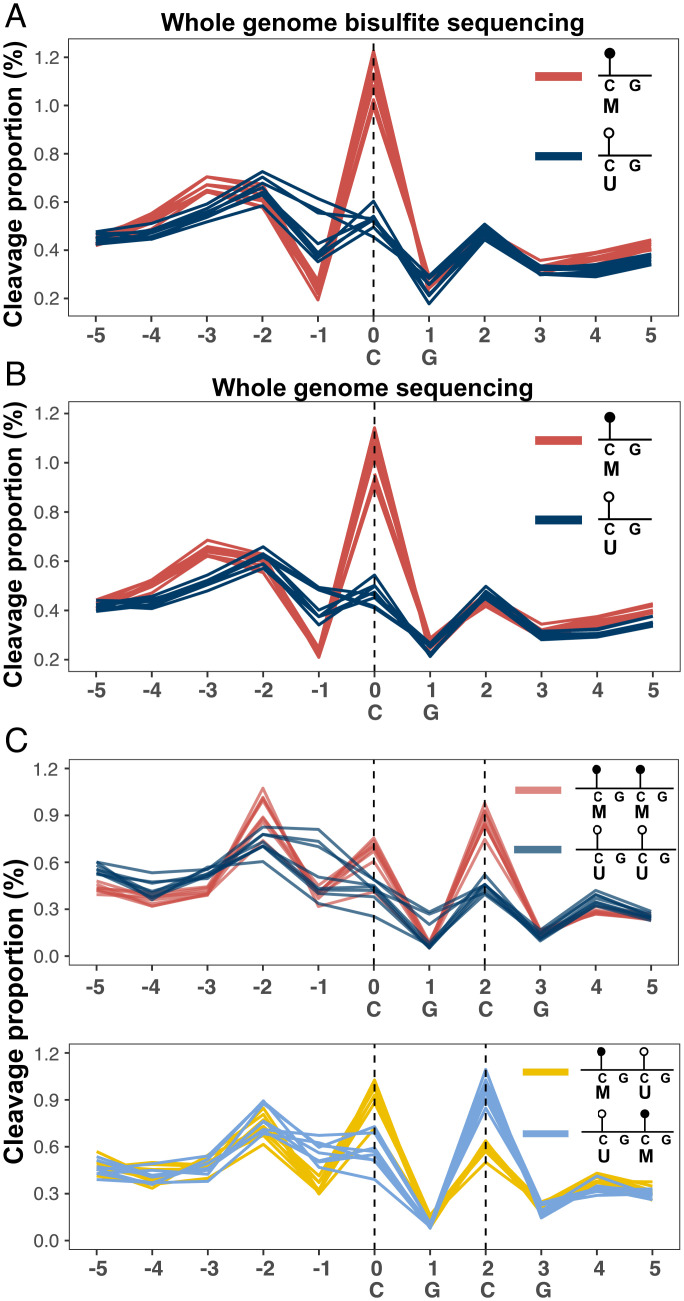
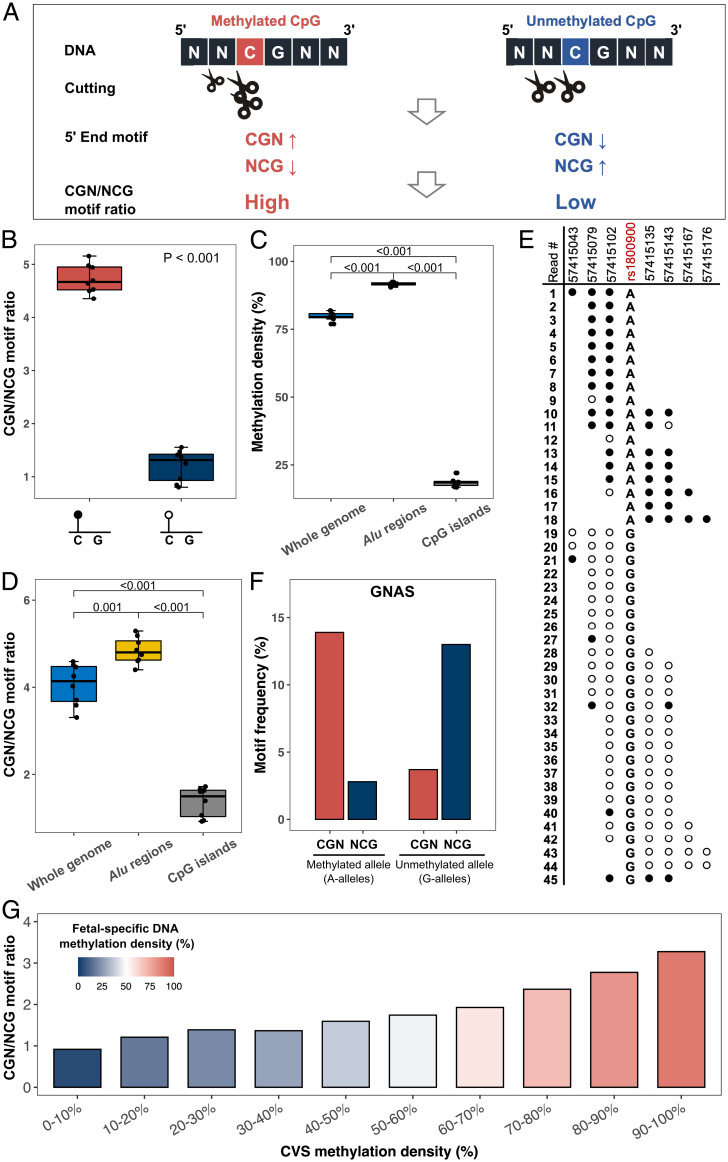
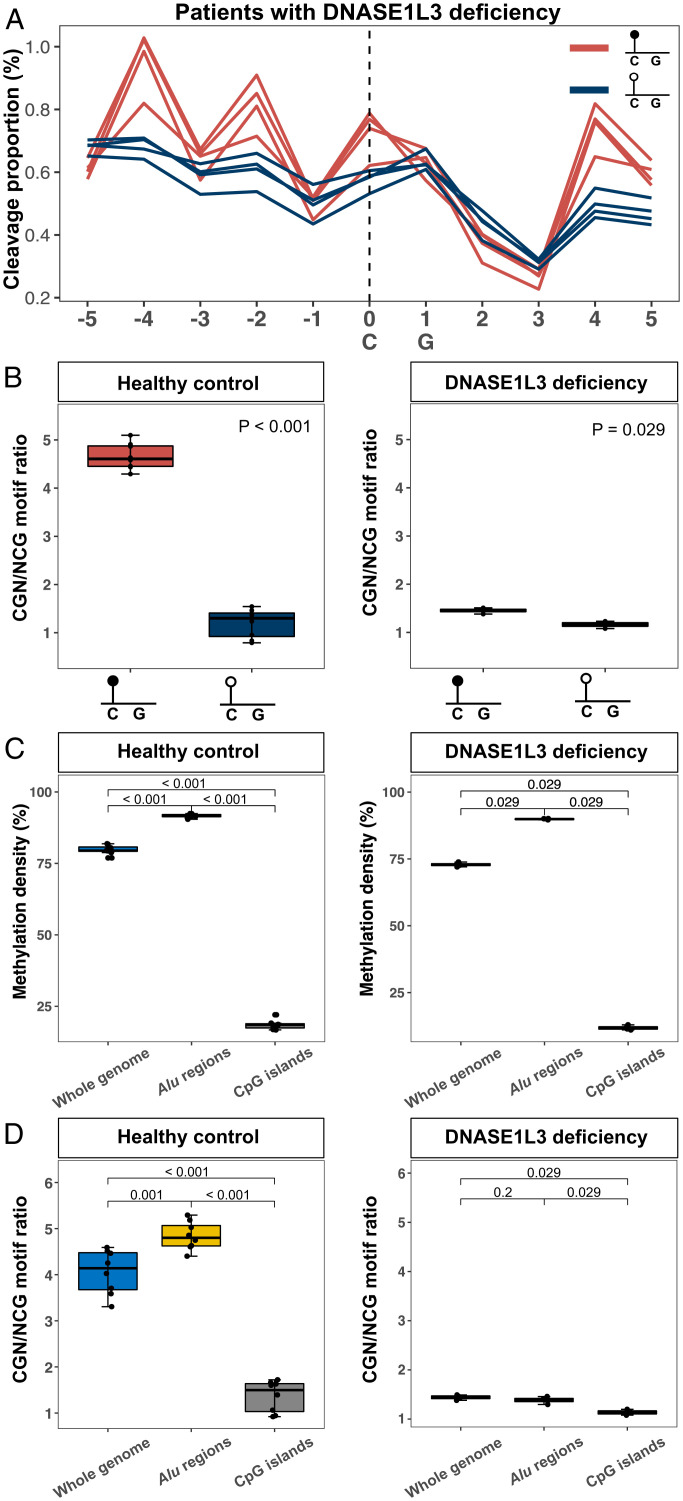
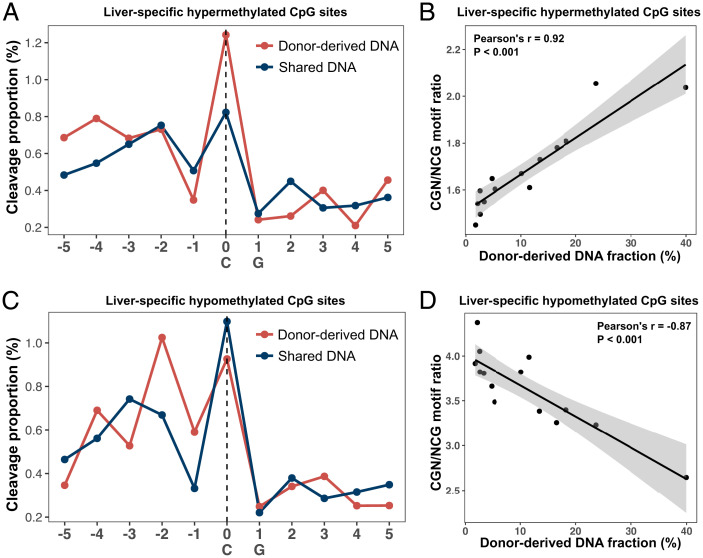
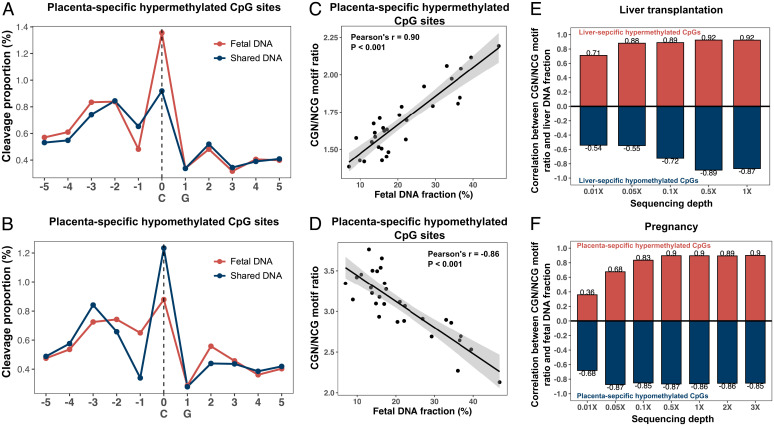
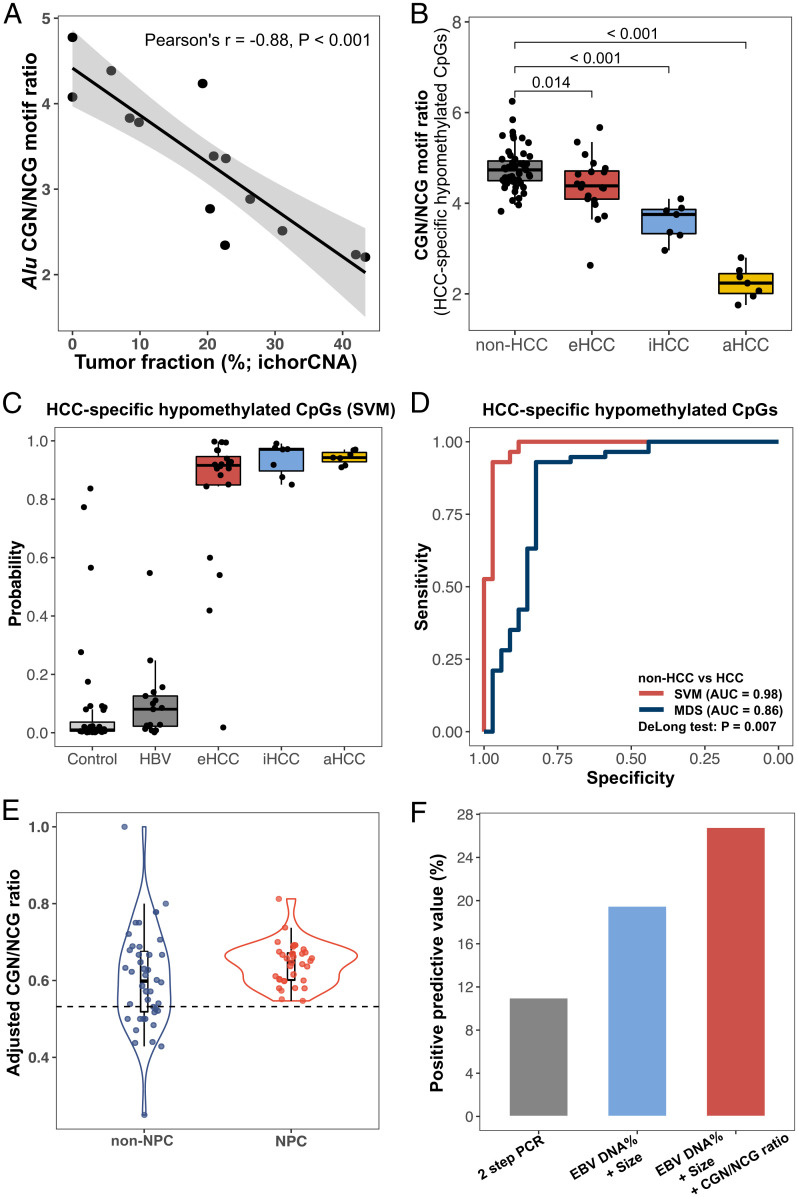
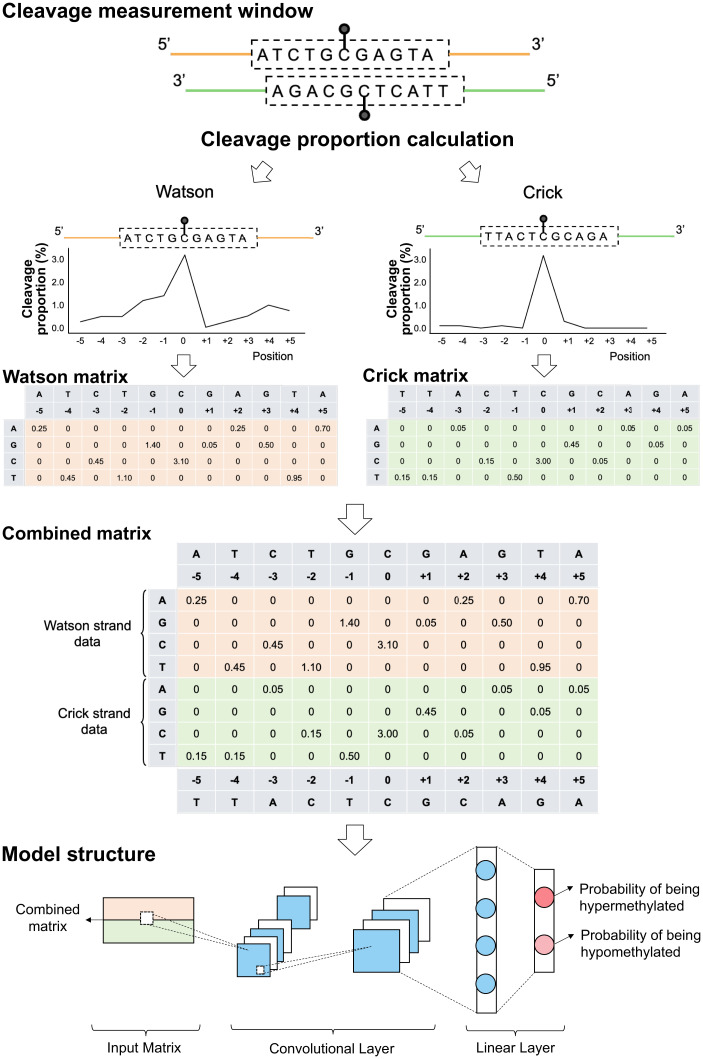
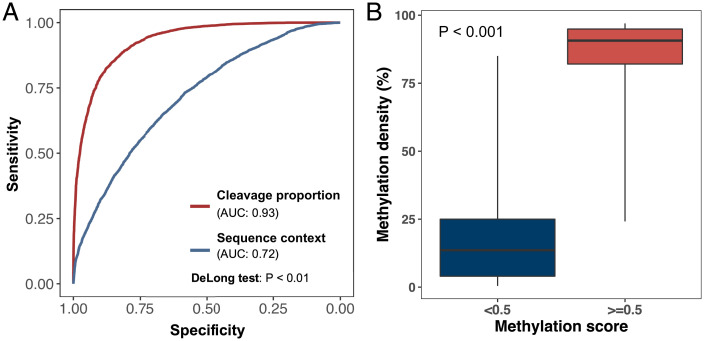
Cell-free DNA (cfDNA) fragmentation patterns contain important molecular information linked to tissues of origin. We explored the possibility of using fragmentation patterns to predict cytosine-phosphate-guanine (CpG) methylation of cfDNA, obviating the use of bisulfite treatment and associated risks of DNA degradation. This study investigated the cfDNA cleavage profile surrounding a CpG (i.e., within an 11-nucleotide [nt] window) to analyze cfDNA methylation. The cfDNA cleavage proportion across positions within the window appeared nonrandom and exhibited correlation with methylation status. The mean cleavage proportion was ∼twofold higher at the cytosine of methylated CpGs than unmethylated ones in healthy controls. In contrast, the mean cleavage proportion rapidly decreased at the 1-nt position immediately preceding methylated CpGs. Such differential cleavages resulted in a characteristic change in relative presentations of CGN and NCG motifs at 5' ends, where N represented any nucleotide. CGN/NCG motif ratios were correlated with methylation levels at tissue-specific methylated CpGs (e.g., placenta or liver) (Pearson's absolute r > 0.86). cfDNA cleavage profiles were thus informative for cfDNA methylation and tissue-of-origin analyses. Using CG-containing end motifs, we achieved an area under a receiver operating characteristic curve (AUC) of 0.98 in differentiating patients with and without hepatocellular carcinoma and enhanced the positive predictive value of nasopharyngeal carcinoma screening (from 19.6 to 26.8%). Furthermore, we elucidated the feasibility of using cfDNA cleavage patterns to deduce CpG methylation at single CpG resolution using a deep learning algorithm and achieved an AUC of 0.93. FRAGmentomics-based Methylation Analysis (FRAGMA) presents many possibilities for noninvasive prenatal, cancer, and organ transplantation assessment.
Keywords: cancer detection; epigenetics; fragmentomics; liquid biopsy; noninvasive prenatal testing.
Conflict of interest statement
Competing interest statement: A patent application on the described technology has been filed by Q.Z., G.K., P.J., R.Q., L.J., R.W.K.C., K.C.A.C., and Y.M.D.L.
Figures
References
-
- Lo Y. M. D., Han D. S. C., Jiang P., Chiu R. W. K., Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 372, eaaw3616 (2021). - PubMed
-
- Lui Y. Y., et al. , Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 48, 421–427 (2002). - PubMed
-
- Lo Y. M., et al. , Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
