

Microbial Dysregulation of the Gut-Lung Axis in Bronchiectasis
- PMID: 36288294
- PMCID: PMC10111978
- DOI: 10.1164/rccm.202205-0893OC
Microbial Dysregulation of the Gut-Lung Axis in Bronchiectasis
Abstract
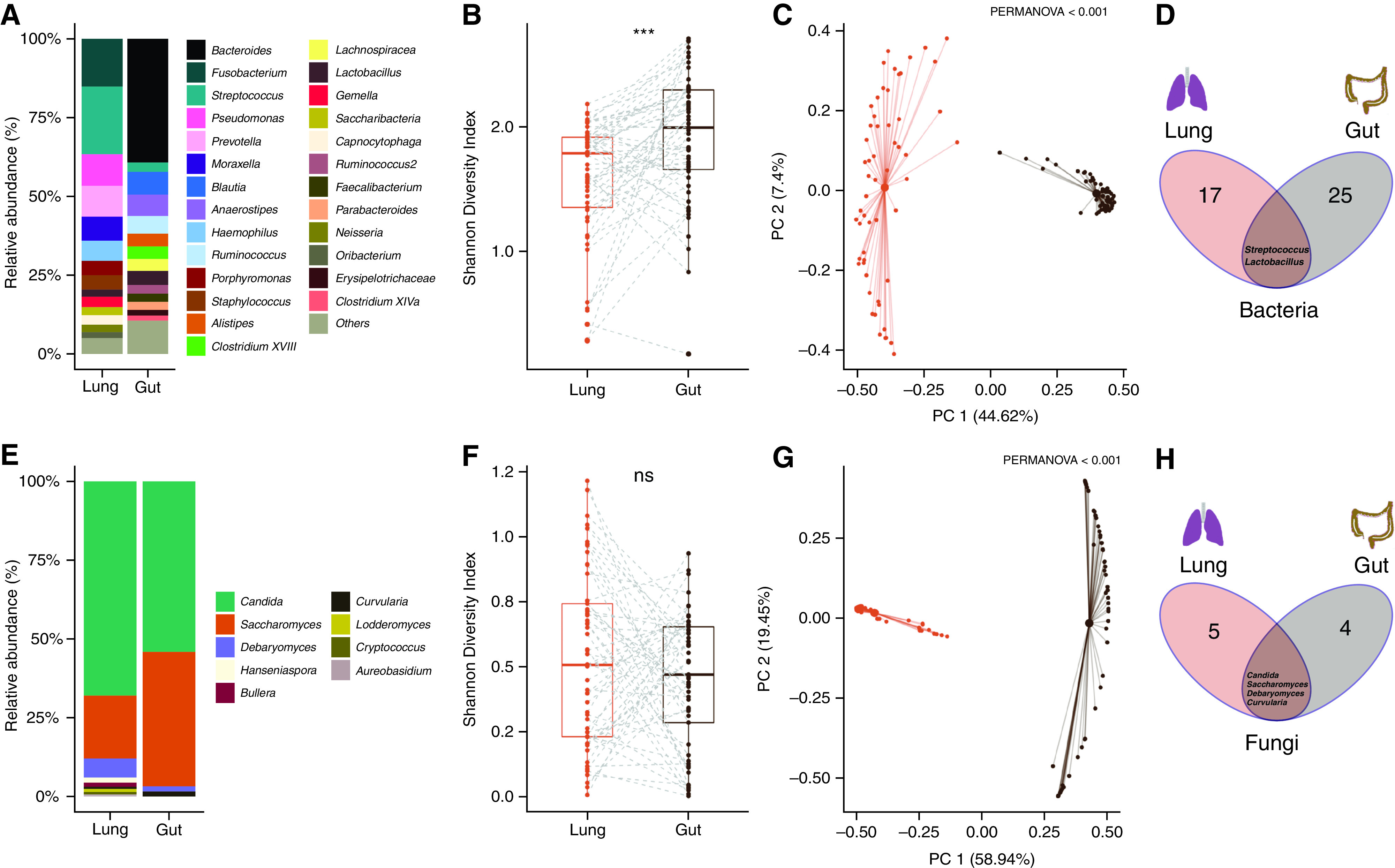
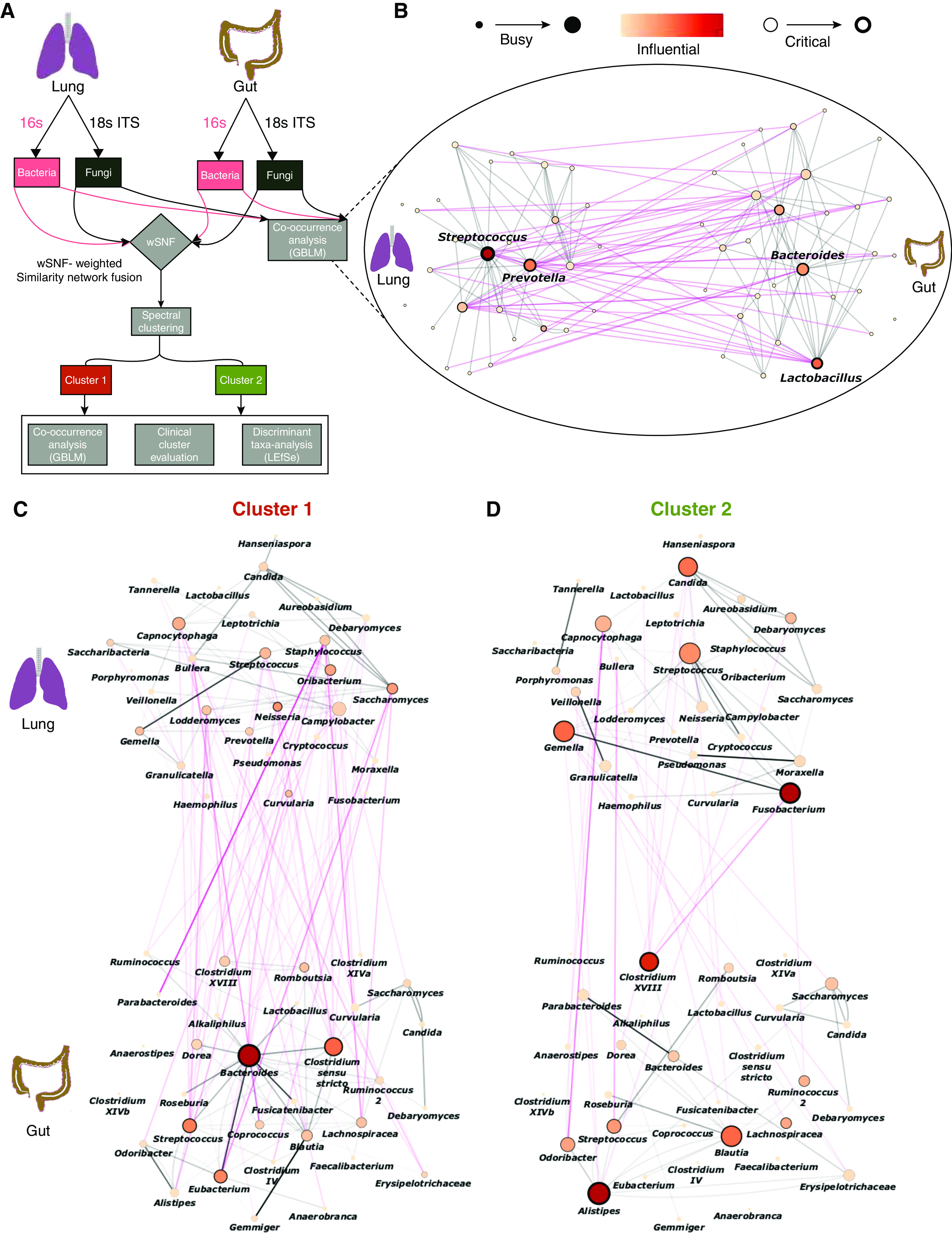
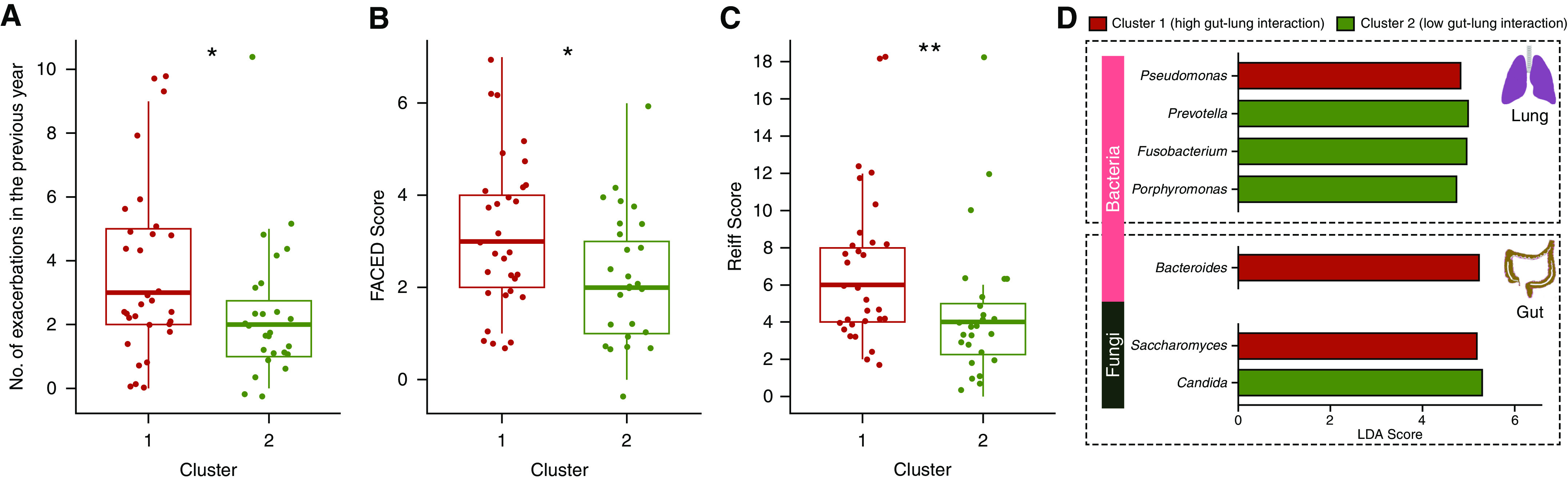
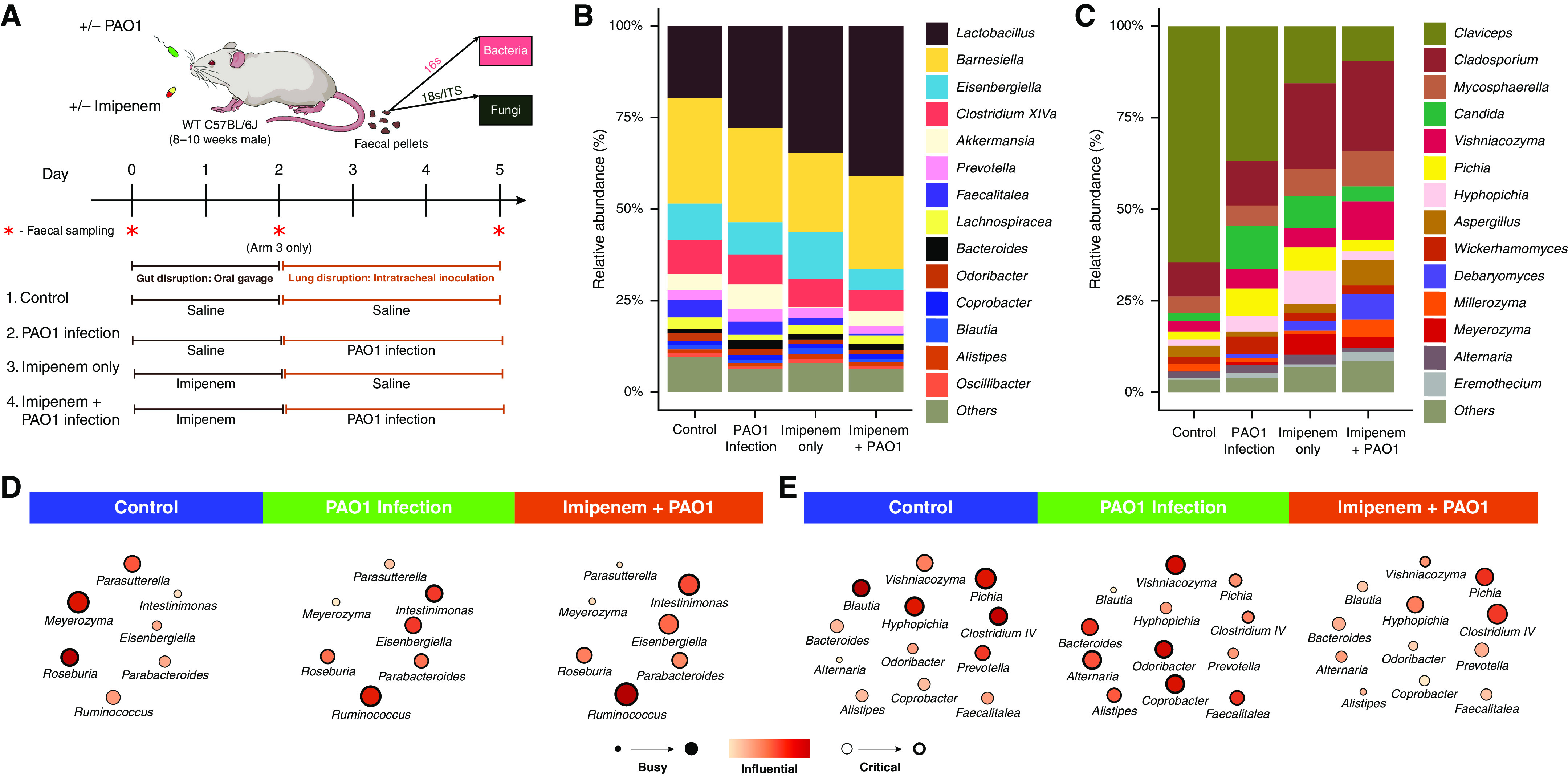
Rationale: Emerging data support the existence of a microbial "gut-lung" axis that remains unexplored in bronchiectasis. Methods: Prospective and concurrent sampling of gut (stool) and lung (sputum) was performed in a cohort of n = 57 individuals with bronchiectasis and subjected to bacteriome (16S rRNA) and mycobiome (18S Internal Transcribed Spacer) sequencing (total, 228 microbiomes). Shotgun metagenomics was performed in a subset (n = 15; 30 microbiomes). Data from gut and lung compartments were integrated by weighted similarity network fusion, clustered, and subjected to co-occurrence analysis to evaluate gut-lung networks. Murine experiments were undertaken to validate specific Pseudomonas-driven gut-lung interactions. Results: Microbial communities in stable bronchiectasis demonstrate a significant gut-lung interaction. Multibiome integration followed by unsupervised clustering reveals two patient clusters, differing by gut-lung interactions and with contrasting clinical phenotypes. A high gut-lung interaction cluster, characterized by lung Pseudomonas, gut Bacteroides, and gut Saccharomyces, is associated with increased exacerbations and greater radiological and overall bronchiectasis severity, whereas the low gut-lung interaction cluster demonstrates an overrepresentation of lung commensals, including Prevotella, Fusobacterium, and Porphyromonas with gut Candida. The lung Pseudomonas-gut Bacteroides relationship, observed in the high gut-lung interaction bronchiectasis cluster, was validated in a murine model of lung Pseudomonas aeruginosa infection. This interaction was abrogated after antibiotic (imipenem) pretreatment in mice confirming the relevance and therapeutic potential of targeting the gut microbiome to influence the gut-lung axis. Metagenomics in a subset of individuals with bronchiectasis corroborated our findings from targeted analyses. Conclusions: A dysregulated gut-lung axis, driven by lung Pseudomonas, associates with poorer clinical outcomes in bronchiectasis.
Keywords: bronchiectasis; gut-lung axis; metagenomics; microbiome; mycobiome.
Figures
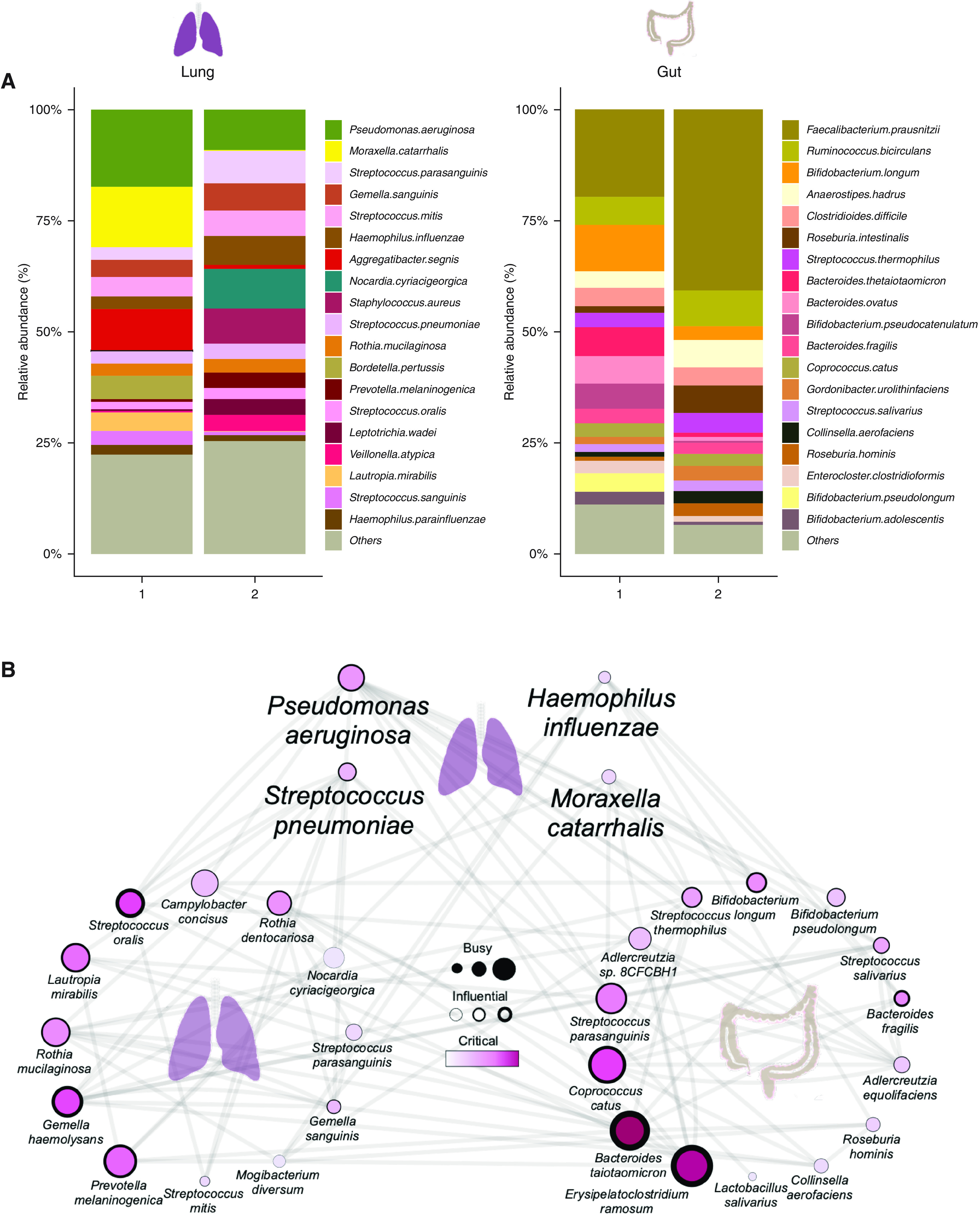
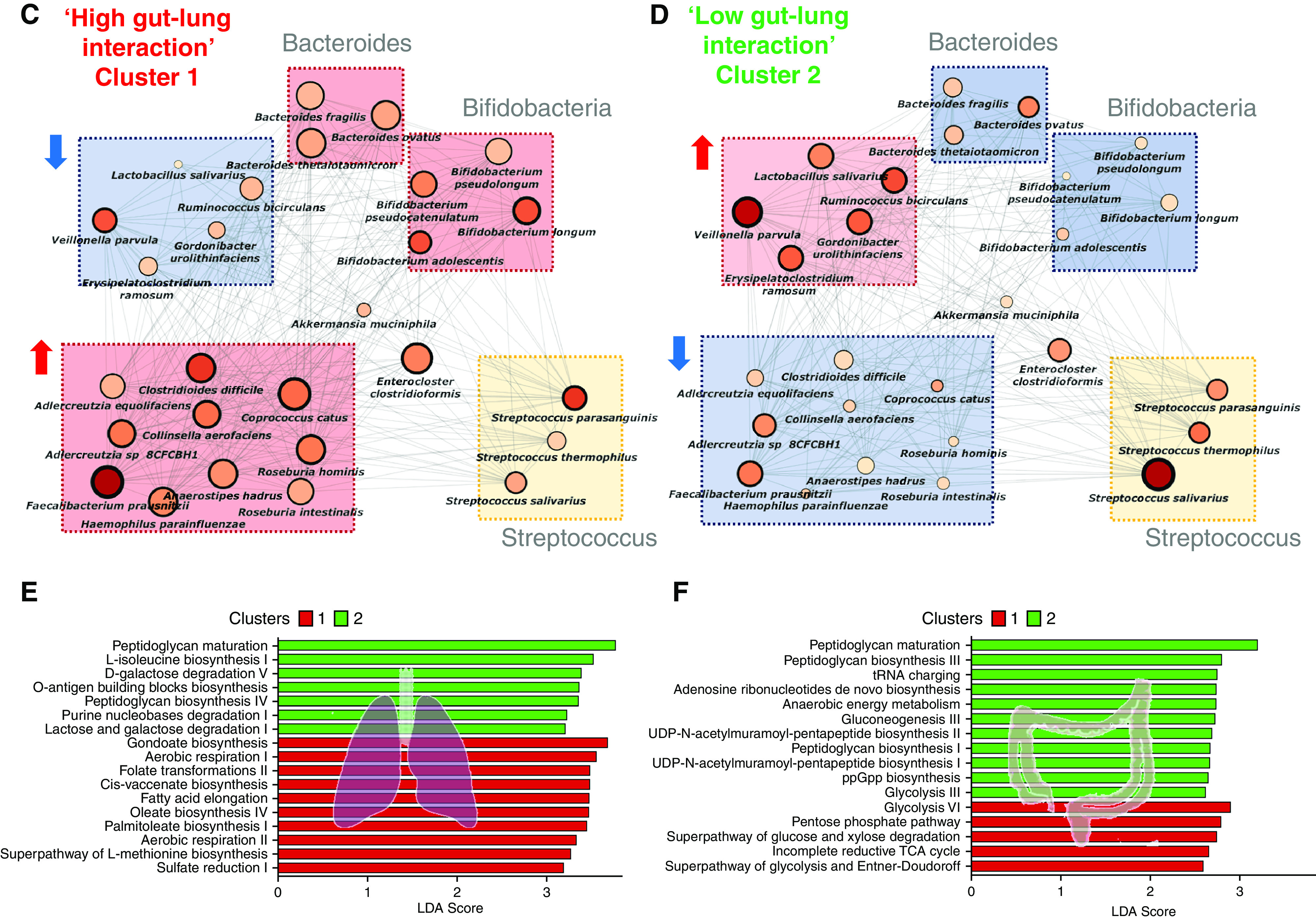
Comment in
-
How to Understand a Revolution: Guts, Lungs, and Bronchiectasis.Am J Respir Crit Care Med. 2023 Apr 1;207(7):812-814. doi: 10.1164/rccm.202211-2179ED. Am J Respir Crit Care Med. 2023. PMID: 36473273 Free PMC article. No abstract available.
-
The Potential Role of Gastric Microbiology in Respiratory Disease.Am J Respir Crit Care Med. 2023 Sep 1;208(5):630-631. doi: 10.1164/rccm.202303-0366LE. Am J Respir Crit Care Med. 2023. PMID: 37348122 Free PMC article. No abstract available.
References
-
- Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ . 2017;356:j831. - PubMed
-
- Tiew PY, Mac Aogáin M, Chotirmall SH. The current understanding and future directions for sputum microbiome profiling in chronic obstructive pulmonary disease. Curr Opin Pulm Med . 2022;28:121–133. - PubMed
