

Control of infection by LC3-associated phagocytosis, CASM, and detection of raised vacuolar pH by the V-ATPase-ATG16L1 axis
- PMID: 36288298
- PMCID: PMC9604538
- DOI: 10.1126/sciadv.abn3298
Control of infection by LC3-associated phagocytosis, CASM, and detection of raised vacuolar pH by the V-ATPase-ATG16L1 axis
Abstract
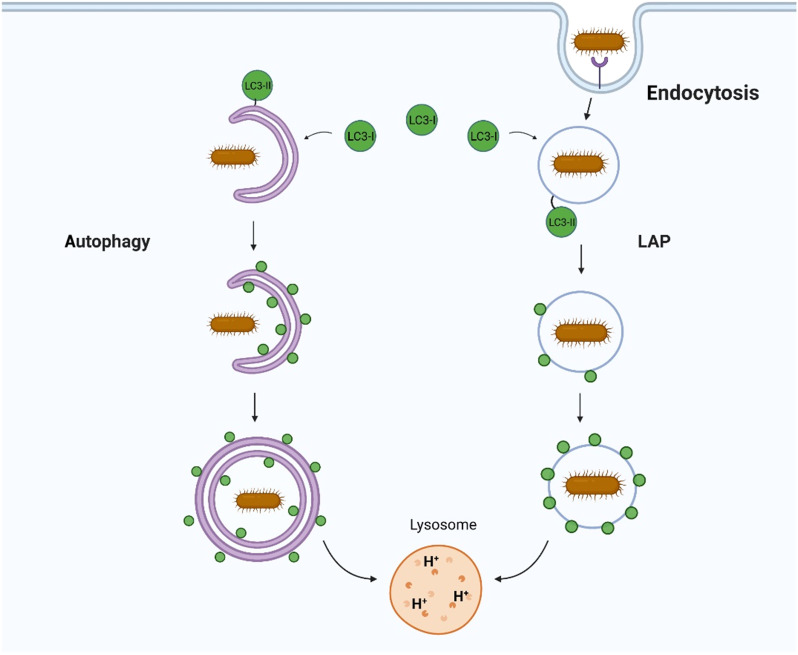
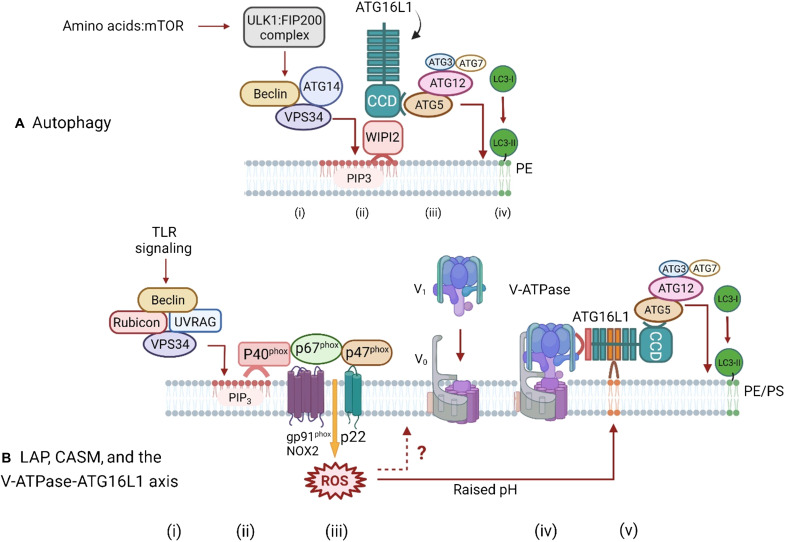
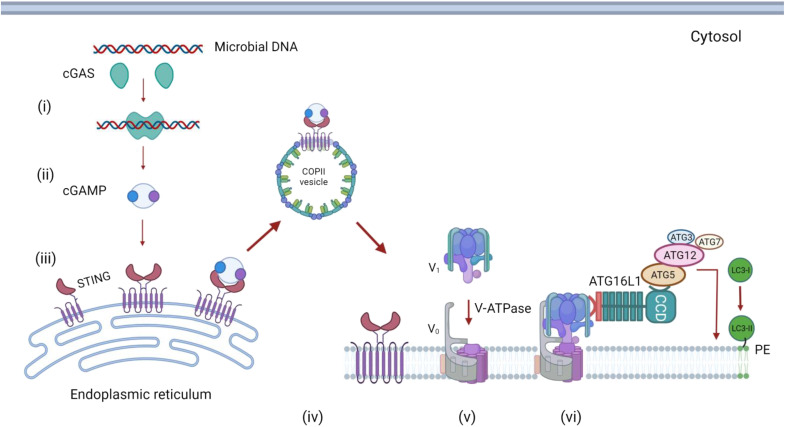
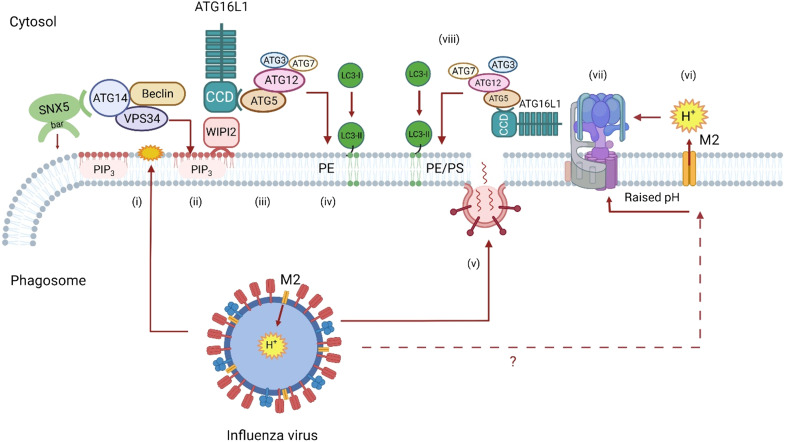
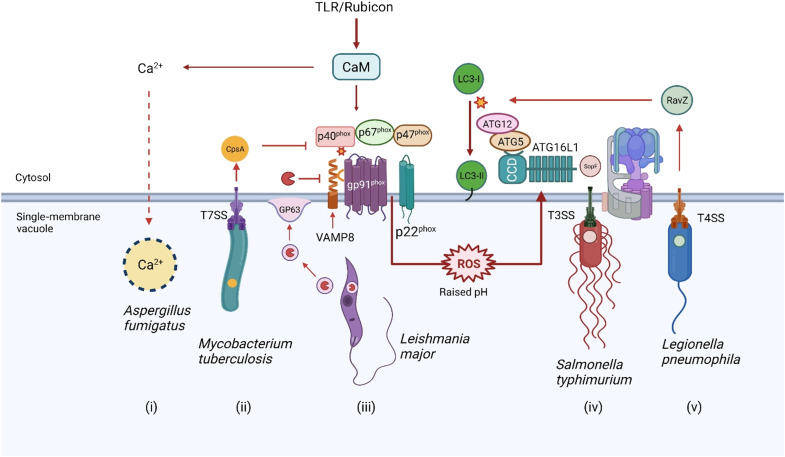
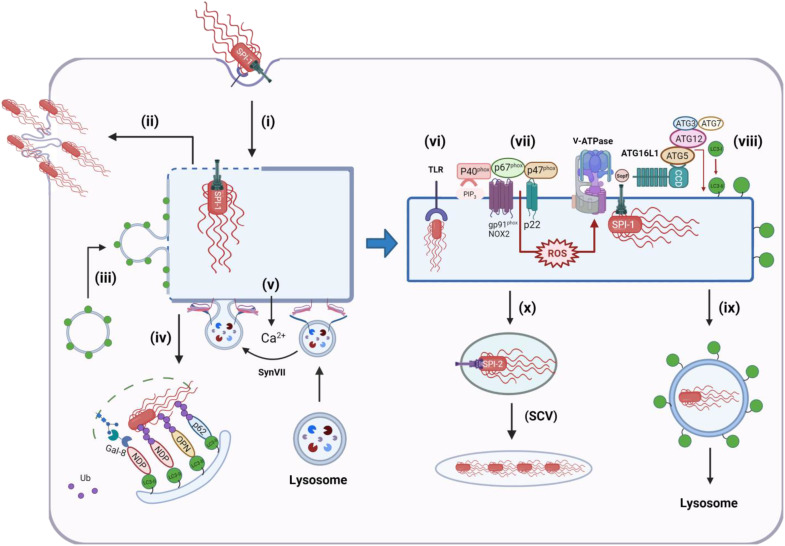
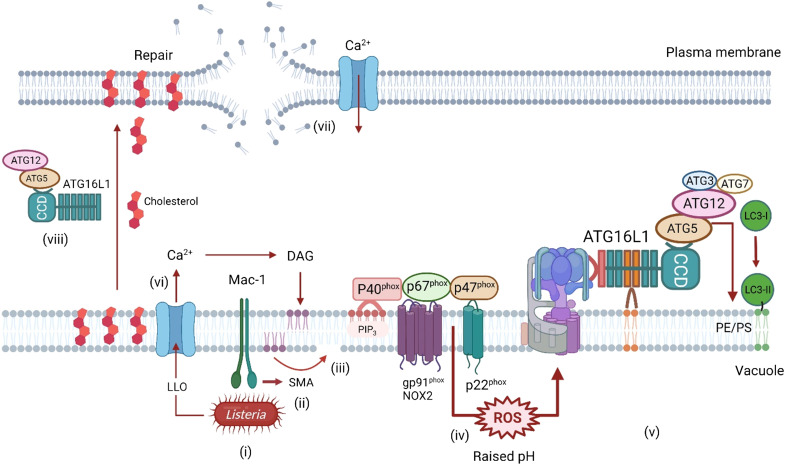
The delivery of pathogens to lysosomes for degradation provides an important defense against infection. Degradation is enhanced when LC3 is conjugated to endosomes and phagosomes containing pathogens to facilitate fusion with lysosomes. In phagocytic cells, TLR signaling and Rubicon activate LC3-associated phagocytosis (LAP) where stabilization of the NADPH oxidase leads to sustained ROS production and raised vacuolar pH. Raised pH triggers the assembly of the vacuolar ATPase on the vacuole membrane where it binds ATG16L1 to recruit the core LC3 conjugation complex (ATG16L1:ATG5-12). This V-ATPase-ATG16L1 axis is also activated in nonphagocytic cells to conjugate LC3 to endosomes containing extracellular microbes. Pathogens provide additional signals for recruitment of LC3 when they raise vacuolar pH with pore-forming toxins and proteins, phospholipases, or specialized secretion systems. Many microbes secrete virulence factors to inhibit ROS production and/or the V-ATPase-ATG16L1 axis to slow LC3 recruitment and avoid degradation in lysosomes.
Figures
References
-
- Mizushima N., A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527 (2018). - PubMed
-
- Sanjuan M. A., Dillon C. P., Tait S. W. G., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J. L., Withoff S., Green D. R., Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 (2007). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
