

Clonal somatic copy number altered driver events inform drug sensitivity in high-grade serous ovarian cancer
- PMID: 36289203
- PMCID: PMC9606297
- DOI: 10.1038/s41467-022-33870-0
Clonal somatic copy number altered driver events inform drug sensitivity in high-grade serous ovarian cancer
Abstract
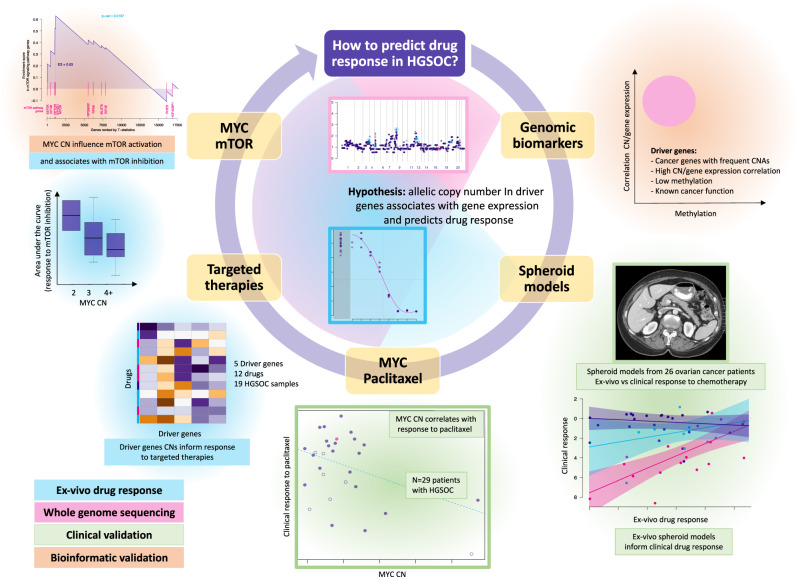
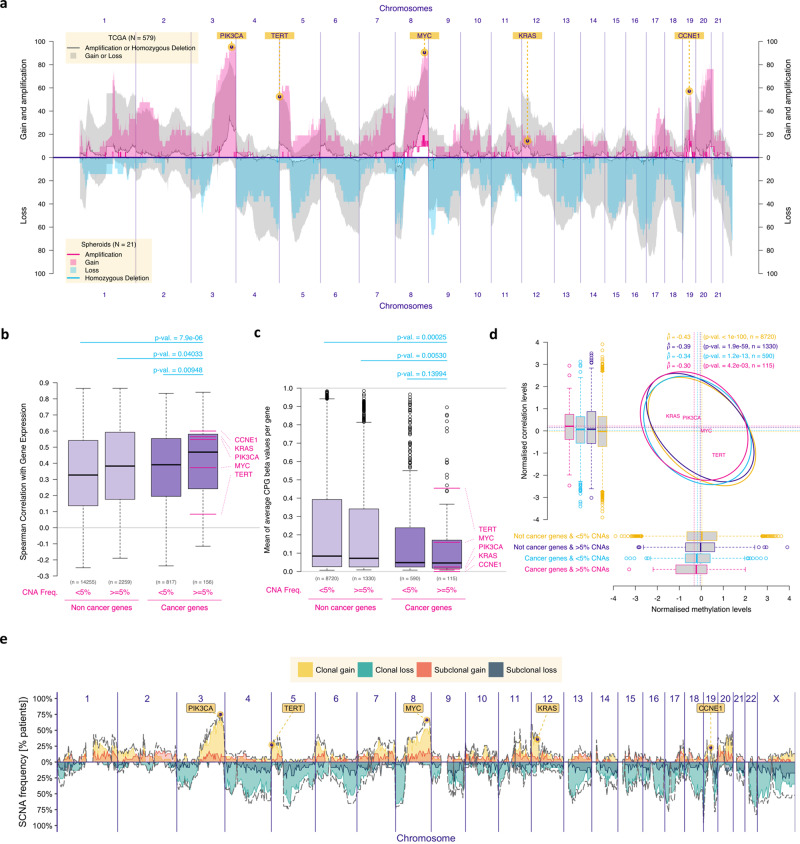
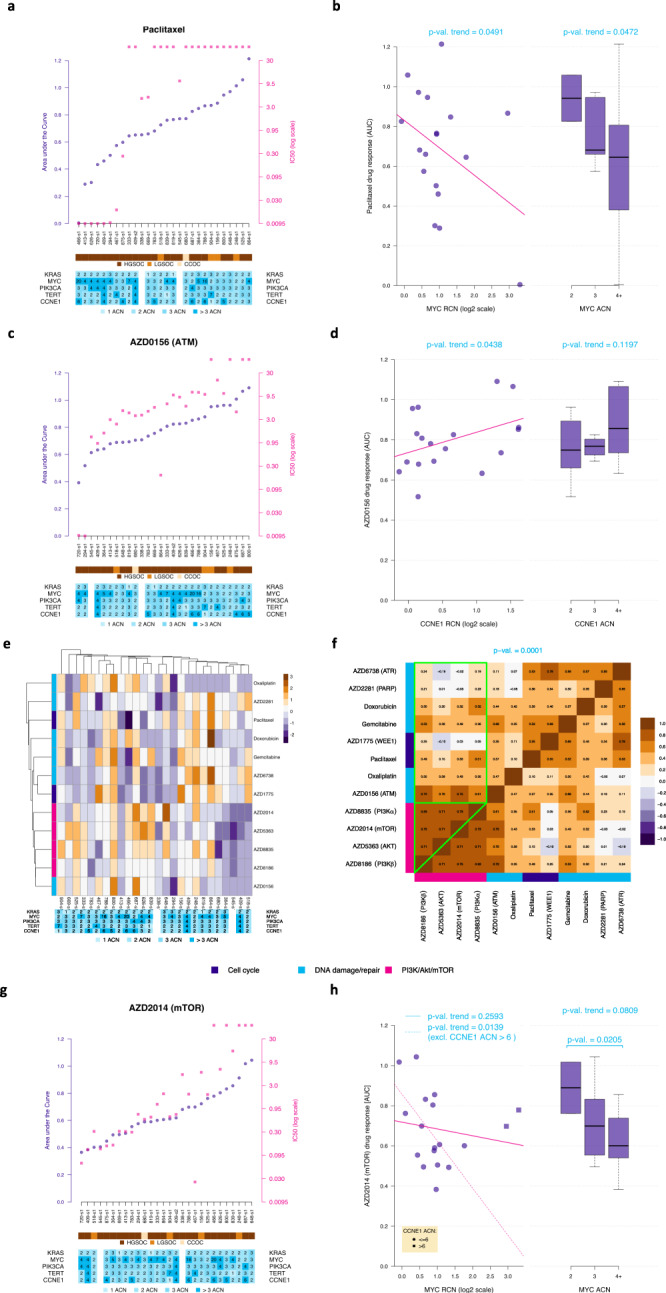
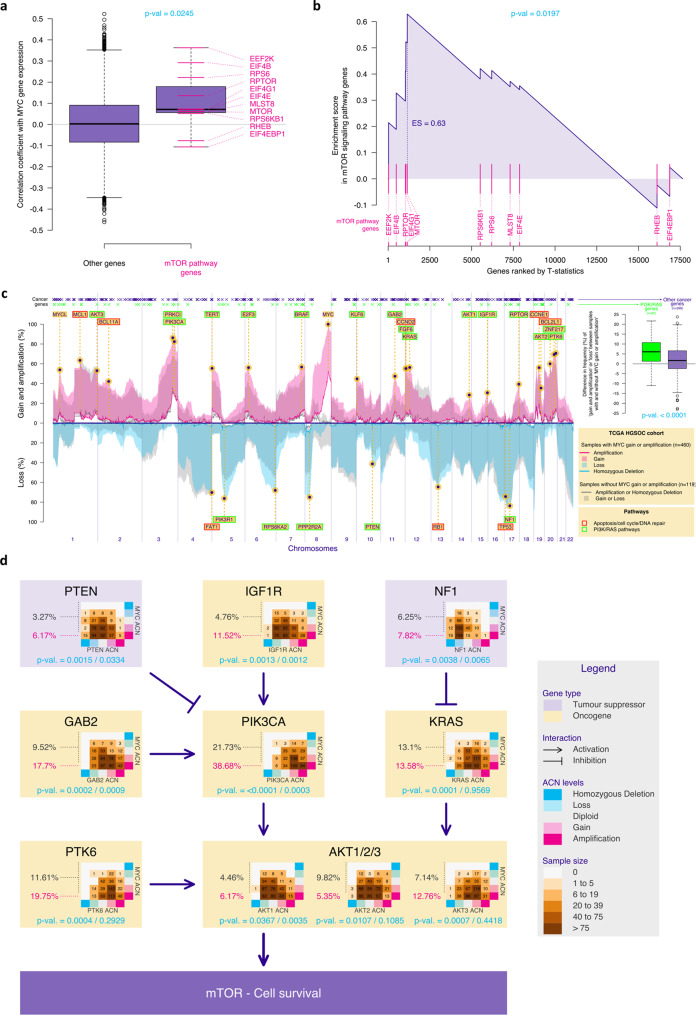
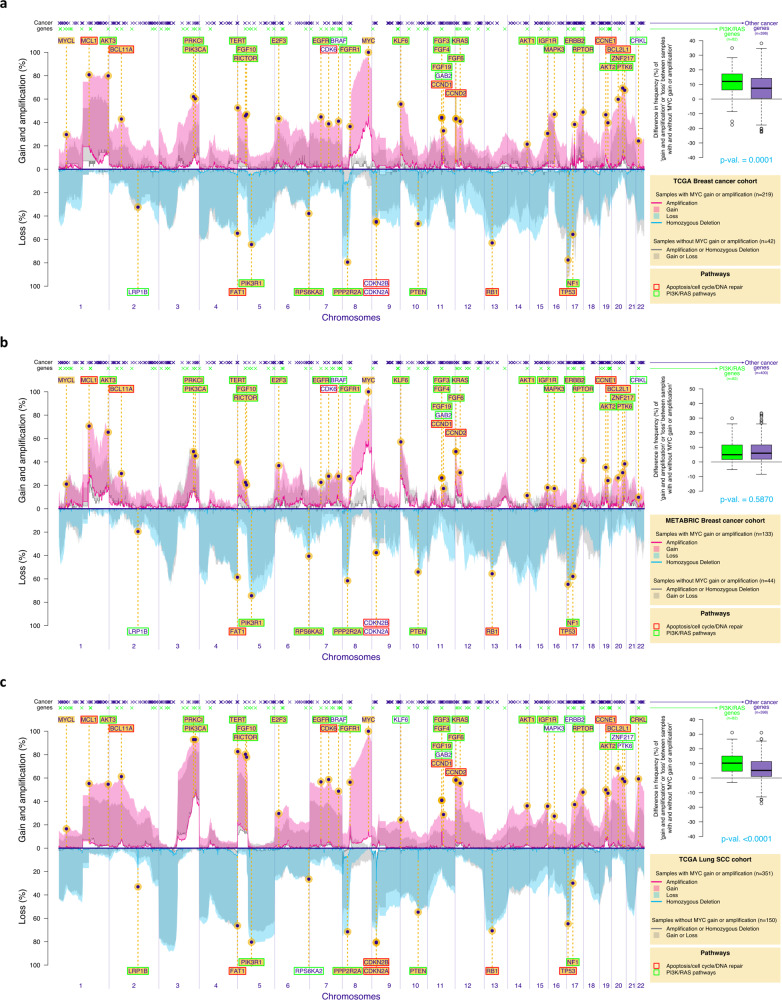
Chromosomal instability is a major challenge to patient stratification and targeted drug development for high-grade serous ovarian carcinoma (HGSOC). Here we show that somatic copy number alterations (SCNAs) in frequently amplified HGSOC cancer genes significantly correlate with gene expression and methylation status. We identify five prevalent clonal driver SCNAs (chromosomal amplifications encompassing MYC, PIK3CA, CCNE1, KRAS and TERT) from multi-regional HGSOC data and reason that their strong selection should prioritise them as key biomarkers for targeted therapies. We use primary HGSOC spheroid models to test interactions between in vitro targeted therapy and SCNAs. MYC chromosomal copy number is associated with in-vitro and clinical response to paclitaxel and in-vitro response to mTORC1/2 inhibition. Activation of the mTOR survival pathway in the context of MYC-amplified HGSOC is statistically associated with increased prevalence of SCNAs in genes from the PI3K pathway. Co-occurrence of amplifications in MYC and genes from the PI3K pathway is independently observed in squamous lung cancer and triple negative breast cancer. In this work, we show that identifying co-occurrence of clonal driver SCNA genes could be used to tailor therapeutics for precision medicine.
© 2022. The Author(s).
Conflict of interest statement
The Experimental Medicine Initiative from the University of Cambridge that funded F.C.M. Clinical Lectureship is partly funded by Astrazeneca. C.C. is a member of the AstraZeneca (AZ) External Science Panel, and has research grants from Roche, Genentech, AZ, and Servier that are administered by the University of Cambridge and reports receiving speakers’ bureau honoraria from Illumina. C.S. acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Invitae (previously Archer Dx Inc - collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical. He is an AstraZeneca Advisory Board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (also SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, Roche Innovation Centre – Shanghai, Metabomed (until July 2022), and the Sarah Canon Research Institute C.S. has received honoraria from Amgen, AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Illumina, and Roche-Ventana. C.S. had stock options in Apogen Biotechnologies and GRAIL until June 2021, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. holds patents relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), US patent relating to detecting tumour mutations (PCT/US2017/28013), methods for lung cancer detection (US20190106751A1) and both a European and US patent related to identifying insertion/deletion mutation targets (PCT/GB2018/051892). J.D.B. has stock options in Tailor Bio and Inivata and is co-founder of Tailor Bio. J.D.B. has had consulting and advisory roles in AstraZeneca and Clovis Oncology and has received honoraria from GSK and Astrazeneca. J.D.B. holds patents relating to TAm-Seq v2 method for ctDNA estimation, enhanced detection of target DNA by fragment size analysis and methods for predicting treatment response in cancers. The remaining authors declare no competing interests.
Figures
References
-
- Tutt A, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
