

The co-evolution of the genome and epigenome in colorectal cancer
- PMID: 36289335
- PMCID: PMC9684080
- DOI: 10.1038/s41586-022-05202-1
The co-evolution of the genome and epigenome in colorectal cancer
Abstract
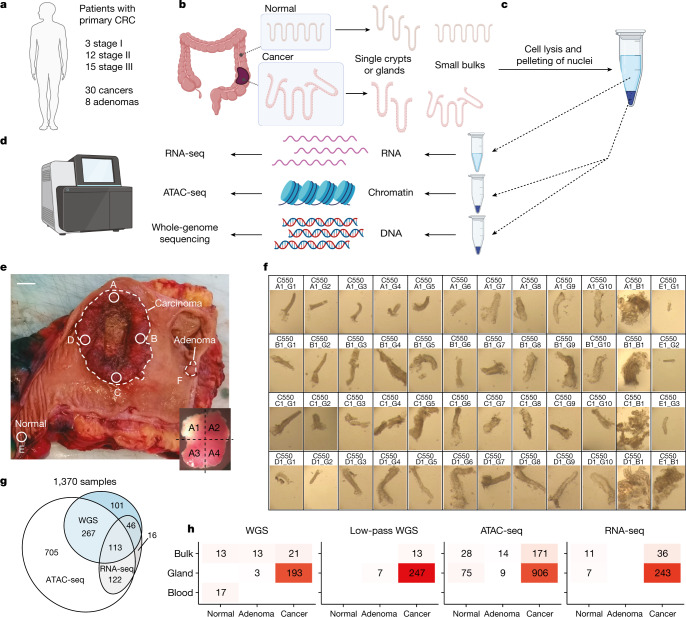
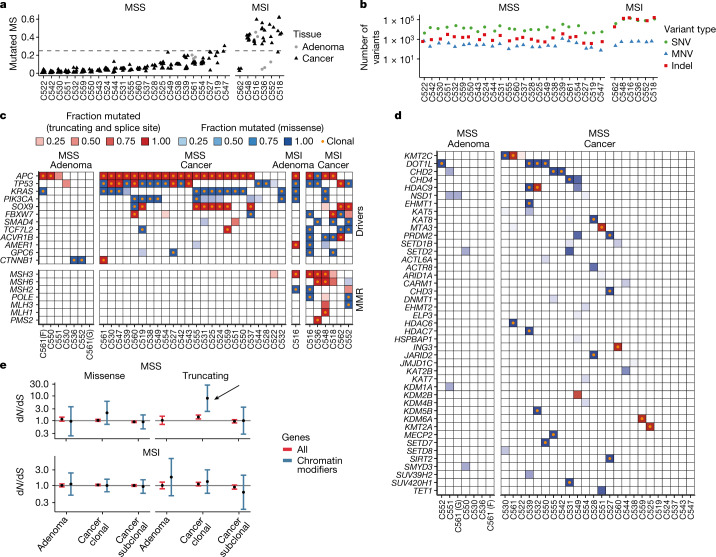
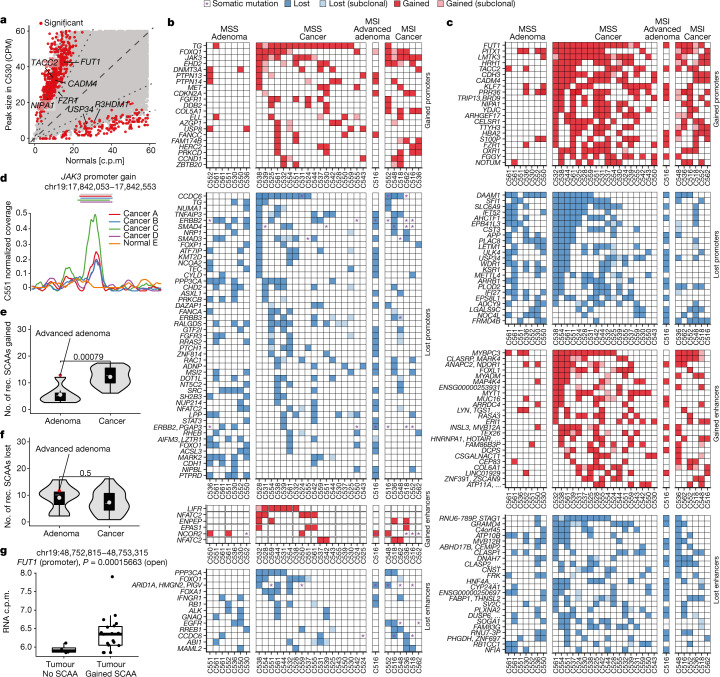
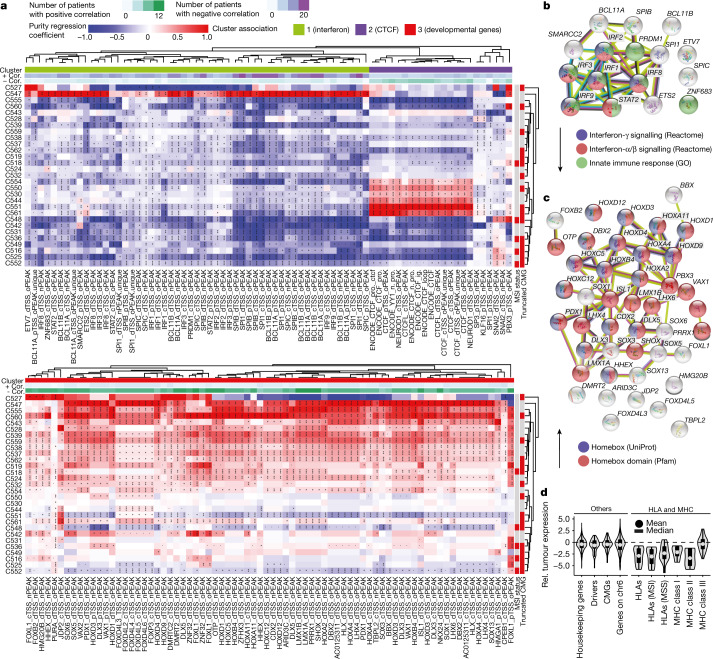
Colorectal malignancies are a leading cause of cancer-related death1 and have undergone extensive genomic study2,3. However, DNA mutations alone do not fully explain malignant transformation4-7. Here we investigate the co-evolution of the genome and epigenome of colorectal tumours at single-clone resolution using spatial multi-omic profiling of individual glands. We collected 1,370 samples from 30 primary cancers and 8 concomitant adenomas and generated 1,207 chromatin accessibility profiles, 527 whole genomes and 297 whole transcriptomes. We found positive selection for DNA mutations in chromatin modifier genes and recurrent somatic chromatin accessibility alterations, including in regulatory regions of cancer driver genes that were otherwise devoid of genetic mutations. Genome-wide alterations in accessibility for transcription factor binding involved CTCF, downregulation of interferon and increased accessibility for SOX and HOX transcription factor families, suggesting the involvement of developmental genes during tumourigenesis. Somatic chromatin accessibility alterations were heritable and distinguished adenomas from cancers. Mutational signature analysis showed that the epigenome in turn influences the accumulation of DNA mutations. This study provides a map of genetic and epigenetic tumour heterogeneity, with fundamental implications for understanding colorectal cancer biology.
© 2022. The Author(s).
Conflict of interest statement
A.-M.B. has received honoraria from Pfizer and Eisai for non-promotional educational content in the field of genomics.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
- 22909/CRUK_/Cancer Research UK/United Kingdom
- 202778/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- P01 CA196569/CA/NCI NIH HHS/United States
- 23110/CRUK_/Cancer Research UK/United Kingdom
- A22909/CRUK_/Cancer Research UK/United Kingdom
- U54 CA217376/CA/NCI NIH HHS/United States
- A23110/CRUK_/Cancer Research UK/United Kingdom
- 105104/Z/14/Z/WT_/Wellcome Trust/United Kingdom
- A19771/CRUK_/Cancer Research UK/United Kingdom
- 19771/CRUK_/Cancer Research UK/United Kingdom
- MR/P000789/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
