

Phenotypic plasticity and genetic control in colorectal cancer evolution
- PMID: 36289336
- PMCID: PMC9684078
- DOI: 10.1038/s41586-022-05311-x
Phenotypic plasticity and genetic control in colorectal cancer evolution
Abstract
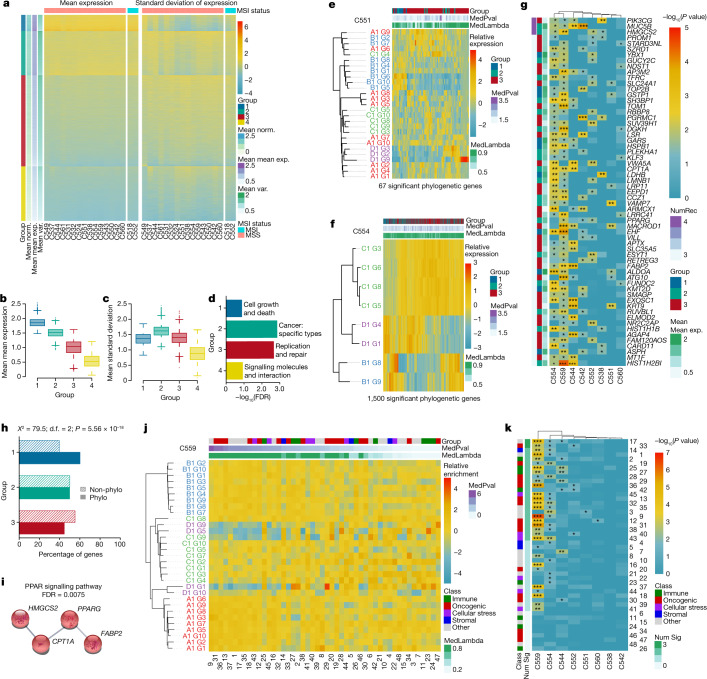
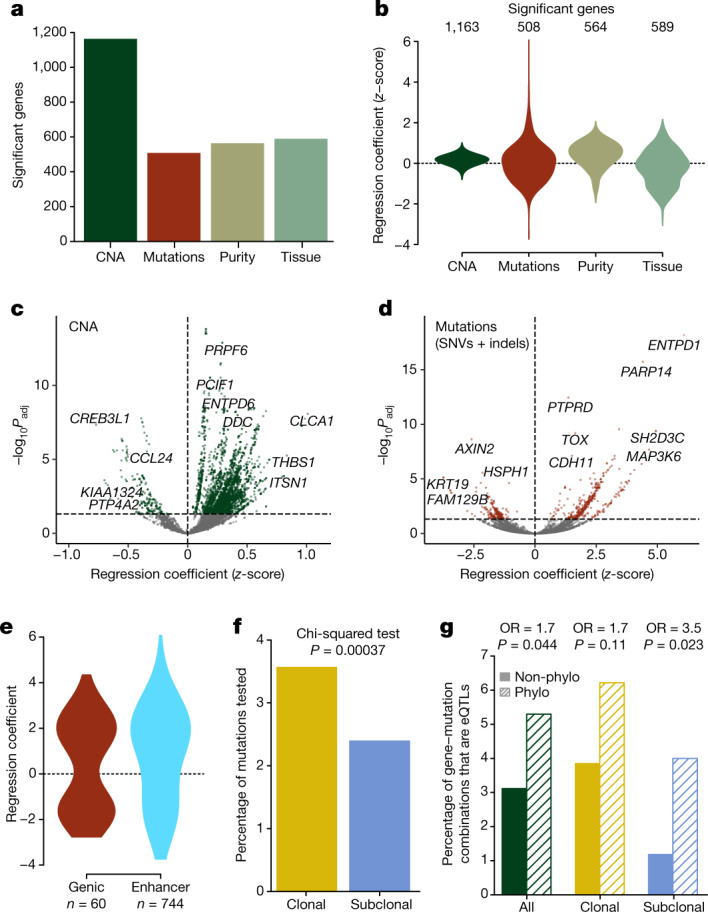
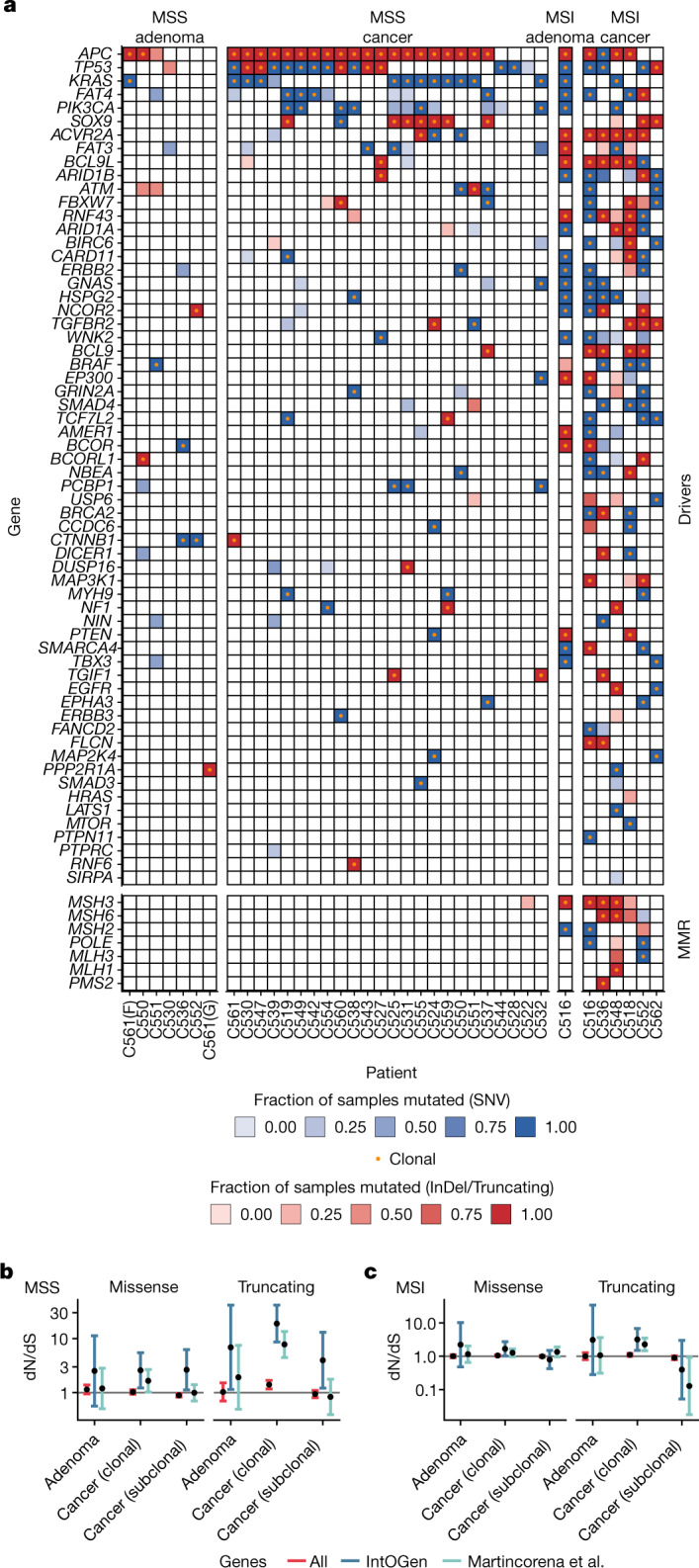
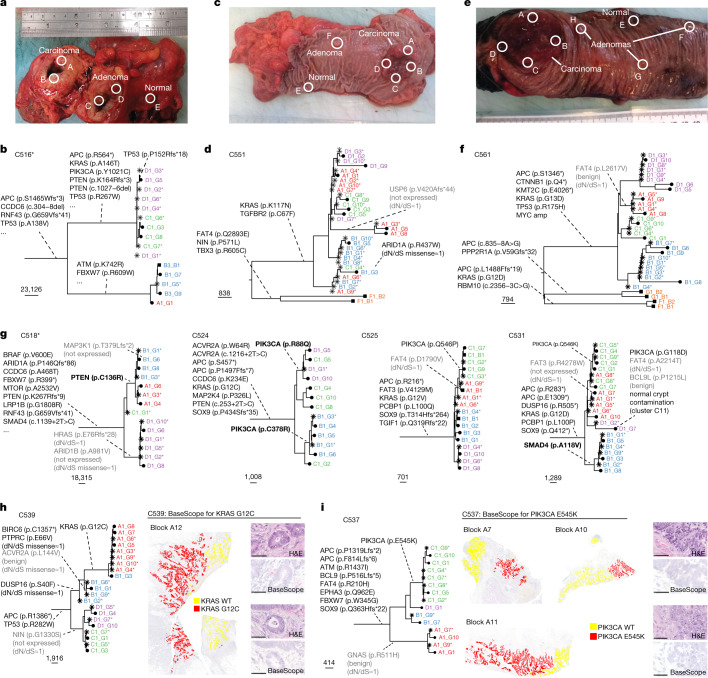
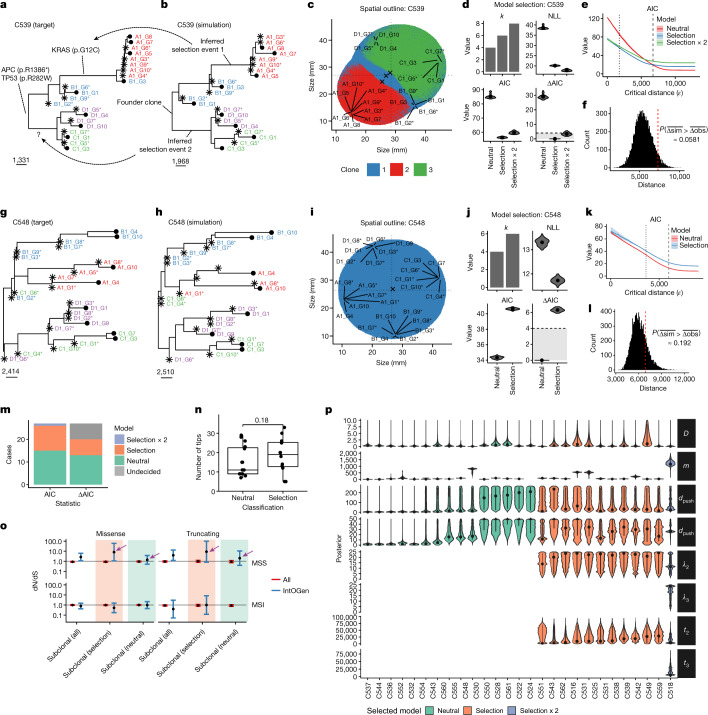
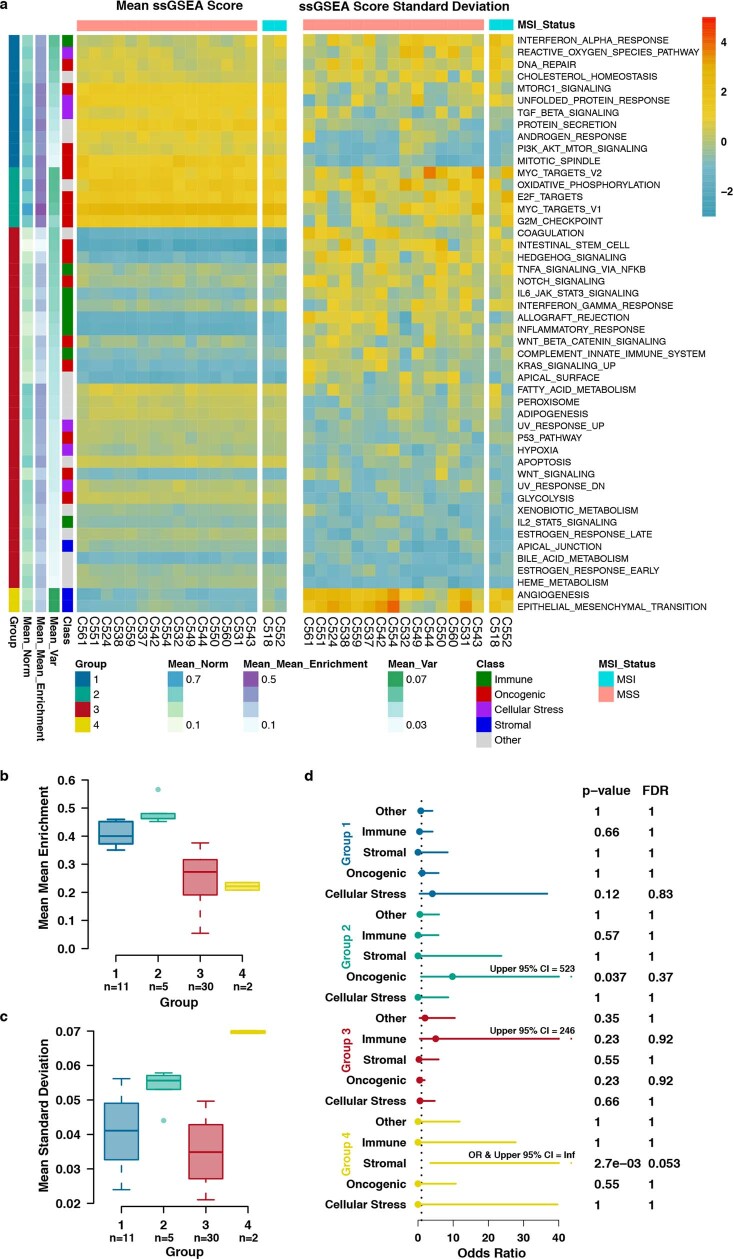
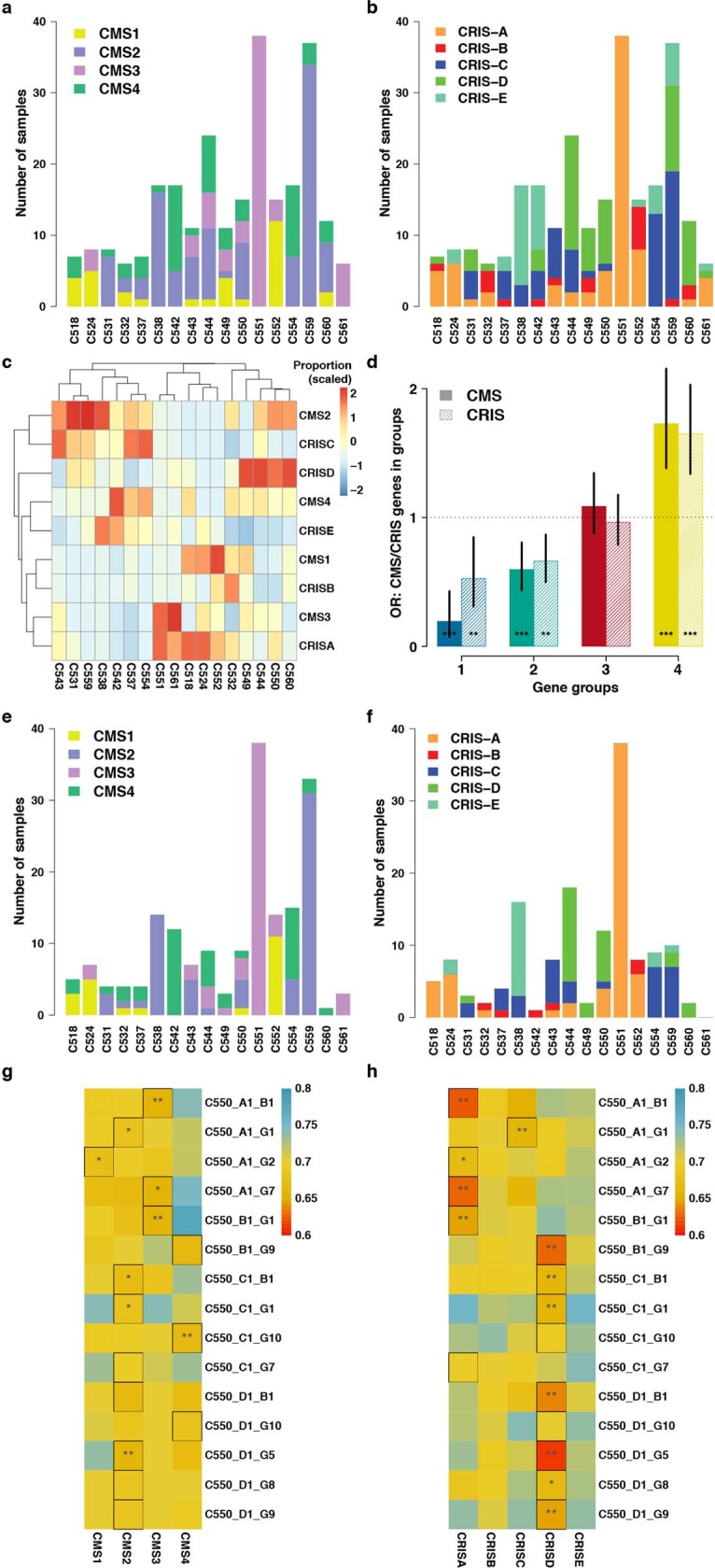
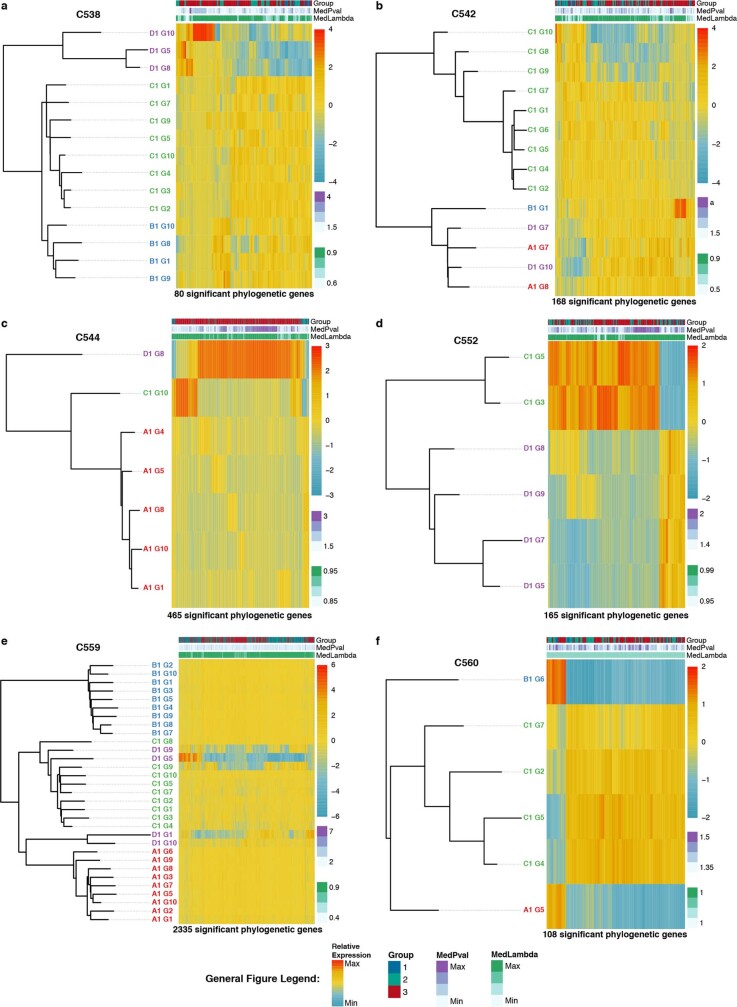
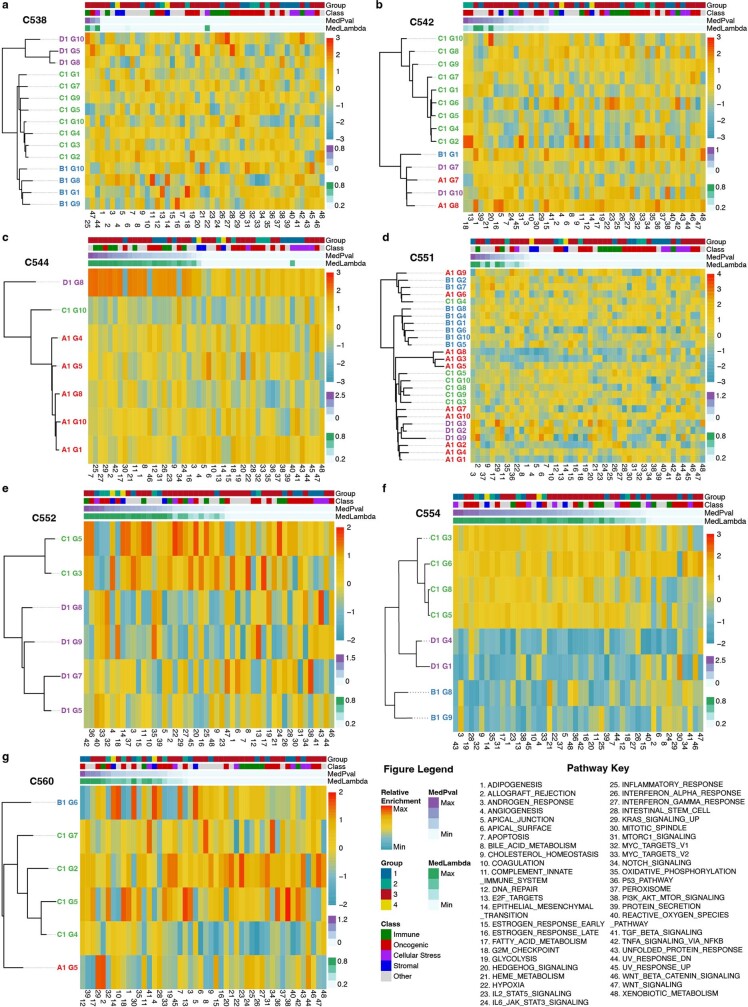
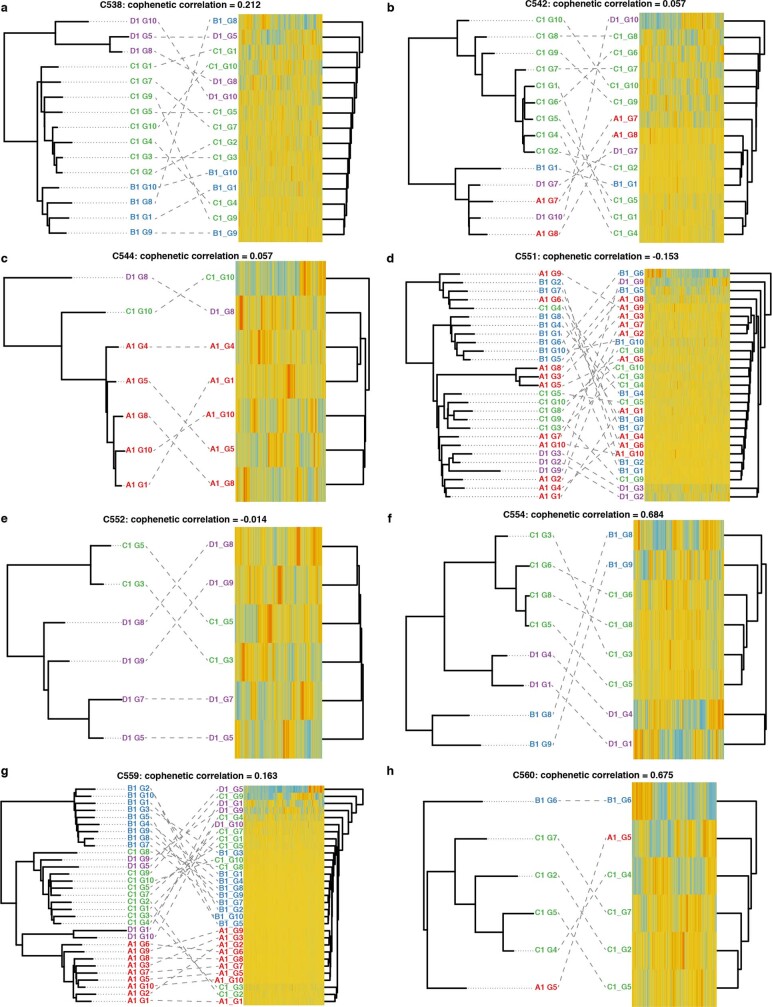
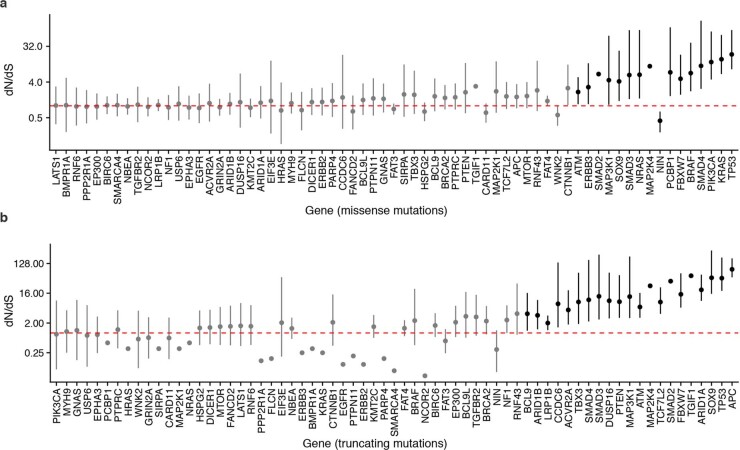
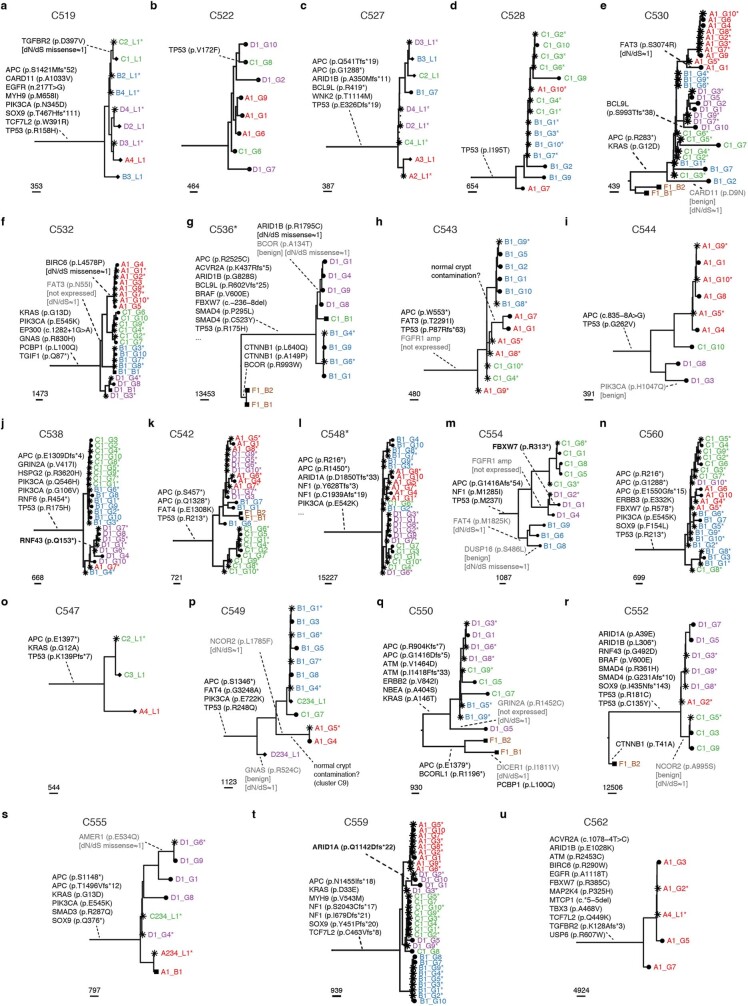
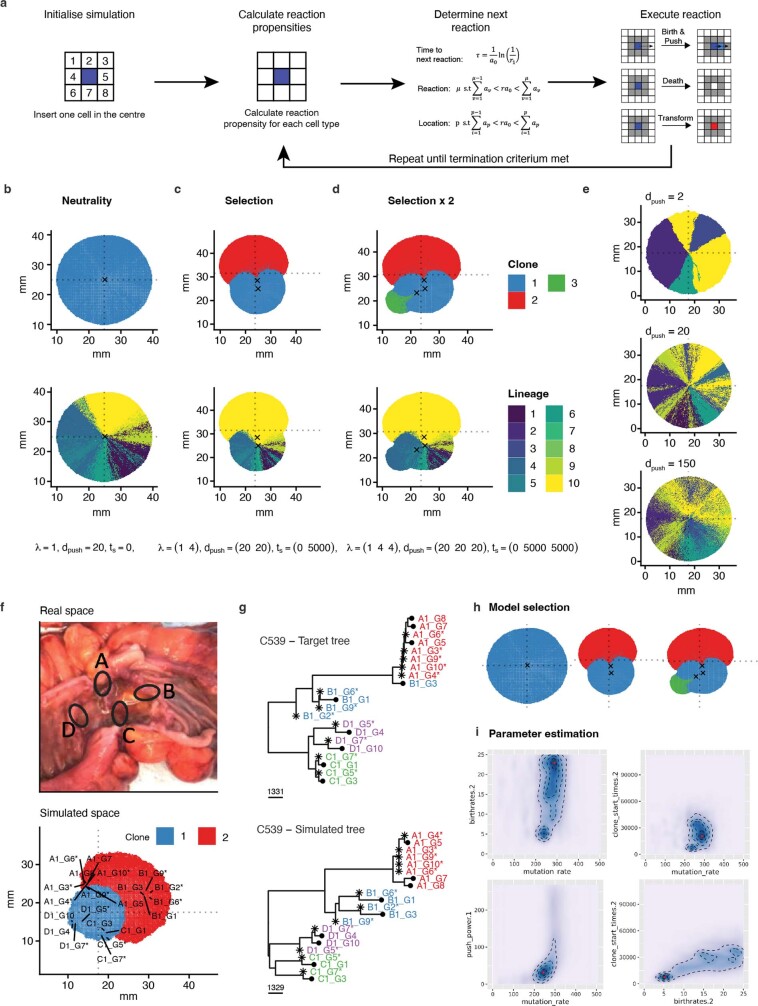
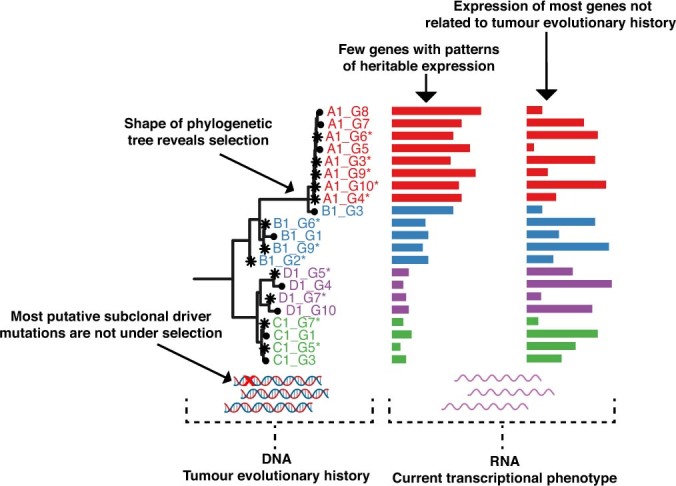
Genetic and epigenetic variation, together with transcriptional plasticity, contribute to intratumour heterogeneity1. The interplay of these biological processes and their respective contributions to tumour evolution remain unknown. Here we show that intratumour genetic ancestry only infrequently affects gene expression traits and subclonal evolution in colorectal cancer (CRC). Using spatially resolved paired whole-genome and transcriptome sequencing, we find that the majority of intratumour variation in gene expression is not strongly heritable but rather 'plastic'. Somatic expression quantitative trait loci analysis identified a number of putative genetic controls of expression by cis-acting coding and non-coding mutations, the majority of which were clonal within a tumour, alongside frequent structural alterations. Consistently, computational inference on the spatial patterning of tumour phylogenies finds that a considerable proportion of CRCs did not show evidence of subclonal selection, with only a subset of putative genetic drivers associated with subclone expansions. Spatial intermixing of clones is common, with some tumours growing exponentially and others only at the periphery. Together, our data suggest that most genetic intratumour variation in CRC has no major phenotypic consequence and that transcriptional plasticity is, instead, widespread within a tumour.
© 2022. The Author(s).
Conflict of interest statement
A.-M.B. has received honoraria from Pfizer and Eisai for non-promotional educational content in the field of genomics. F.I. receives funding from Open Targets, a public–private initiative involving academia and industry, and performs consultancy for the joint CRUK–AstraZeneca Functional Genomics Centre. All other authors declare no competing interests.
Figures
References
-
- Black, J. R. M. & McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer21, 379–392 (2021). - PubMed
Publication types
MeSH terms
Grants and funding
- A26815/CRUK_/Cancer Research UK/United Kingdom
- 22909/CRUK_/Cancer Research UK/United Kingdom
- 202778/B/16/Z/WT_/Wellcome Trust/United Kingdom
- 202778/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- P01 CA196569/CA/NCI NIH HHS/United States
- 23110/CRUK_/Cancer Research UK/United Kingdom
- 209409/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- A22909/CRUK_/Cancer Research UK/United Kingdom
- U54 CA217376/CA/NCI NIH HHS/United States
- 105104/Z/14/Z/WT_/Wellcome Trust/United Kingdom
- A19771/CRUK_/Cancer Research UK/United Kingdom
- 19771/CRUK_/Cancer Research UK/United Kingdom
- MR/P000789/1/MRC_/Medical Research Council/United Kingdom
- 26815/CRUK_/Cancer Research UK/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
