

Divergent clonal differentiation trajectories of T cell exhaustion
- PMID: 36289450
- PMCID: PMC11225711
- DOI: 10.1038/s41590-022-01337-5
Divergent clonal differentiation trajectories of T cell exhaustion
Abstract
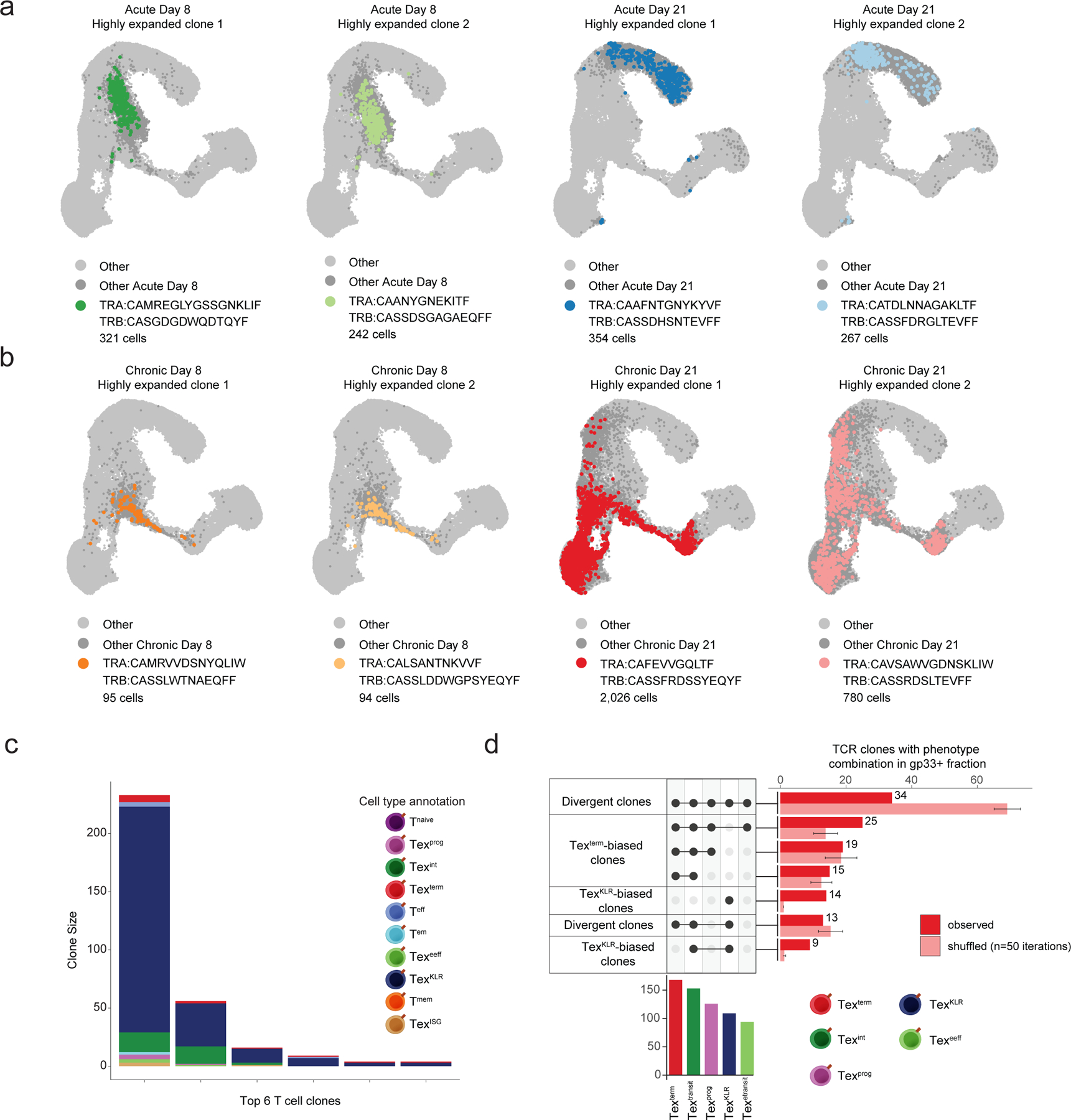
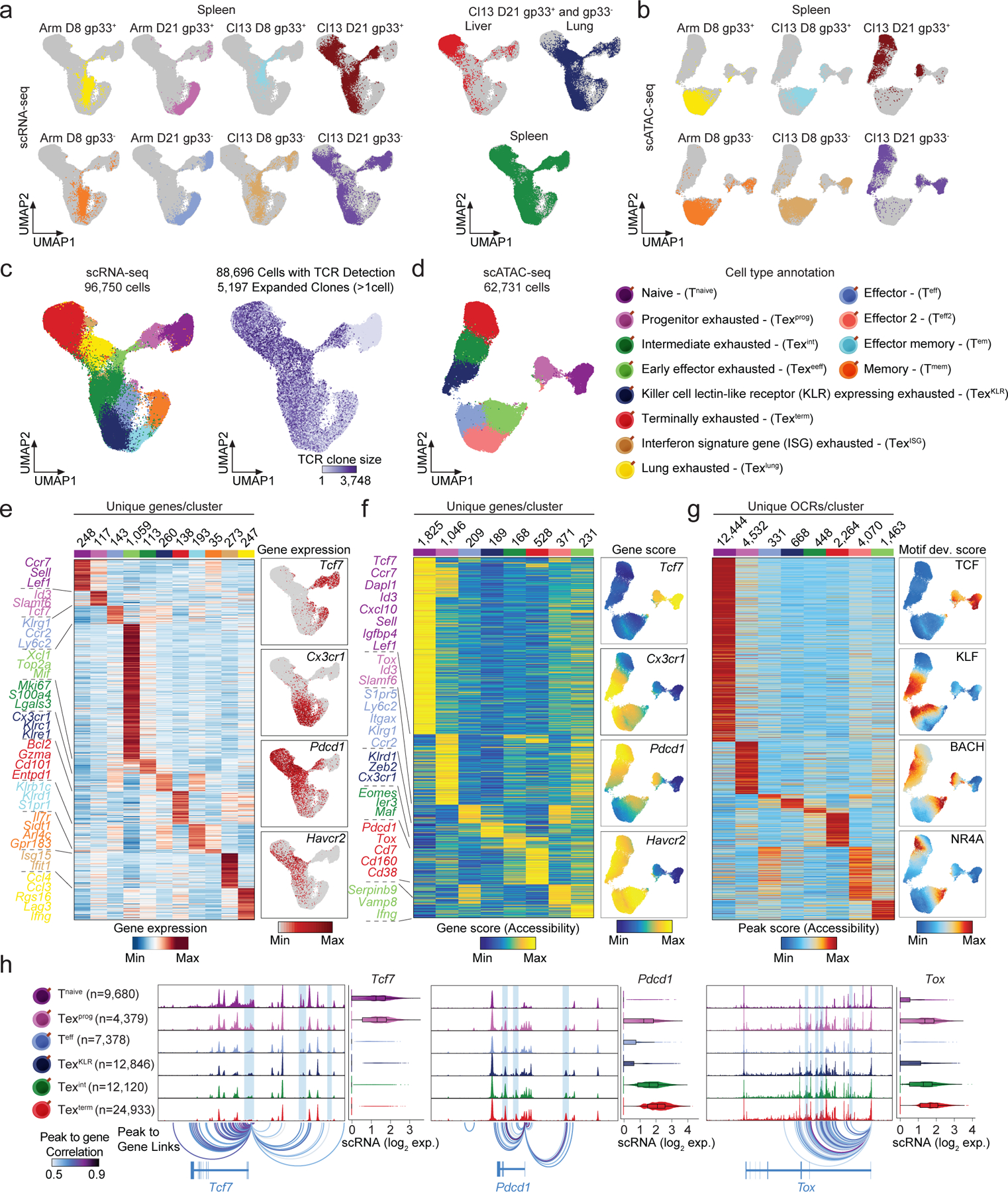
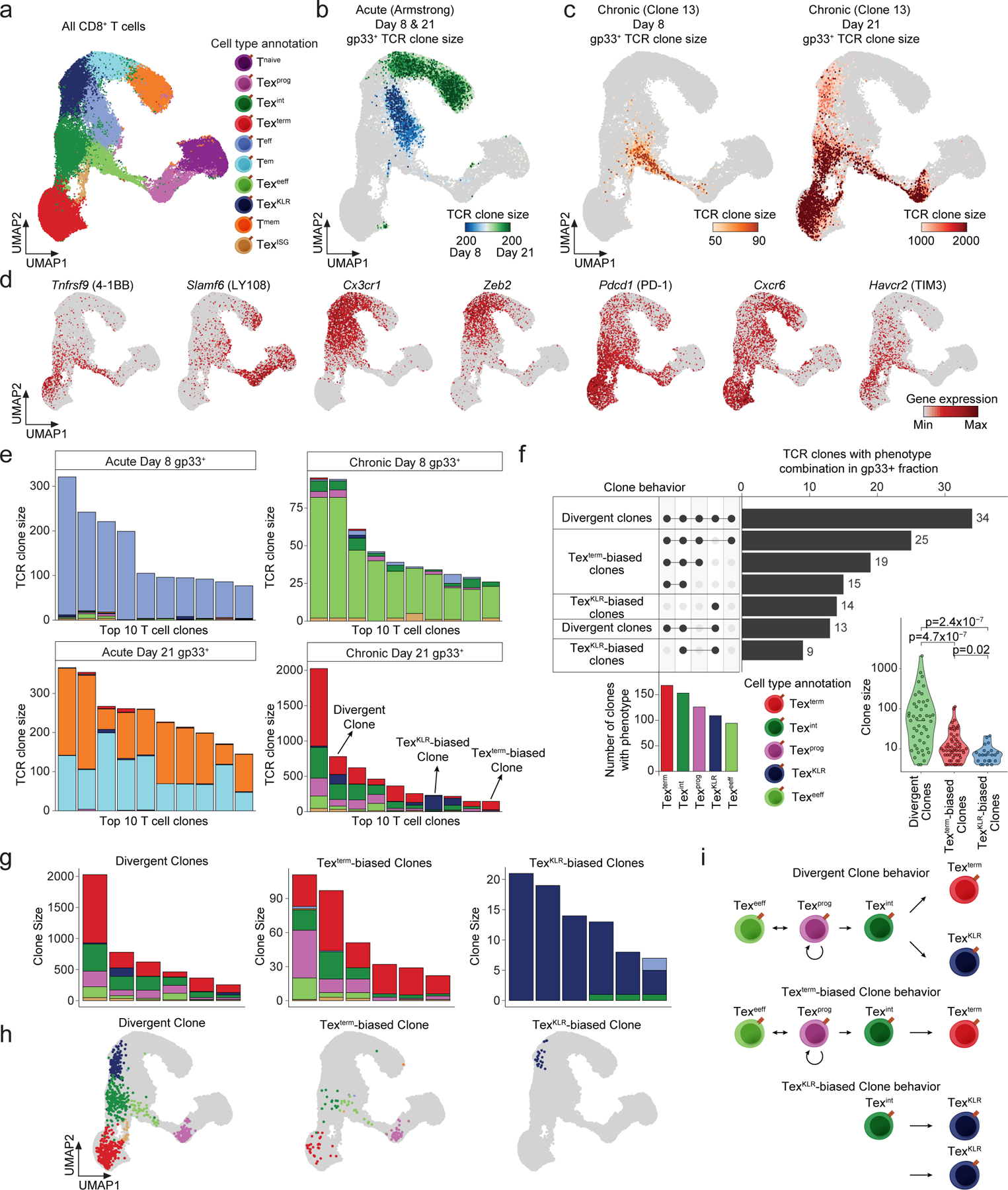
Chronic antigen exposure during viral infection or cancer promotes an exhausted T cell (Tex) state with reduced effector function. However, whether all antigen-specific T cell clones follow the same Tex differentiation trajectory remains unclear. Here, we generate a single-cell multiomic atlas of T cell exhaustion in murine chronic viral infection that redefines Tex phenotypic diversity, including two late-stage Tex subsets with either a terminal exhaustion (Texterm) or a killer cell lectin-like receptor-expressing cytotoxic (TexKLR) phenotype. We use paired single-cell RNA and T cell receptor sequencing to uncover clonal differentiation trajectories of Texterm-biased, TexKLR-biased or divergent clones that acquire both phenotypes. We show that high T cell receptor signaling avidity correlates with Texterm, whereas low avidity correlates with effector-like TexKLR fate. Finally, we identify similar clonal differentiation trajectories in human tumor-infiltrating lymphocytes. These findings reveal clonal heterogeneity in the T cell response to chronic antigen that influences Tex fates and persistence.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures












References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
