

Pathogenesis of Huntington's Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies
- PMID: 36291322
- PMCID: PMC9599635
- DOI: 10.3390/brainsci12101389
Pathogenesis of Huntington's Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies
Abstract
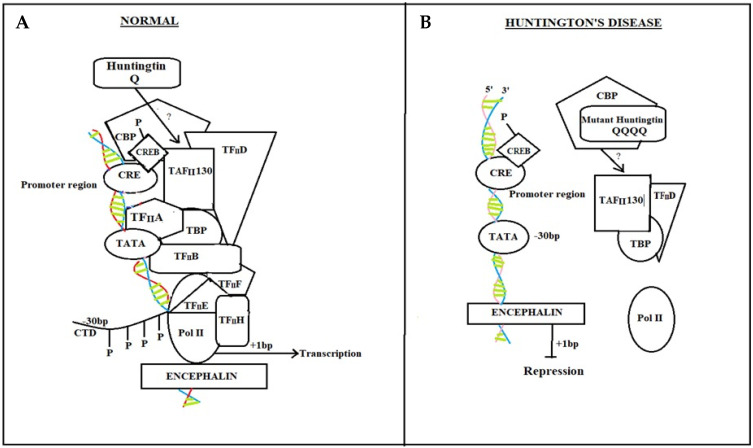
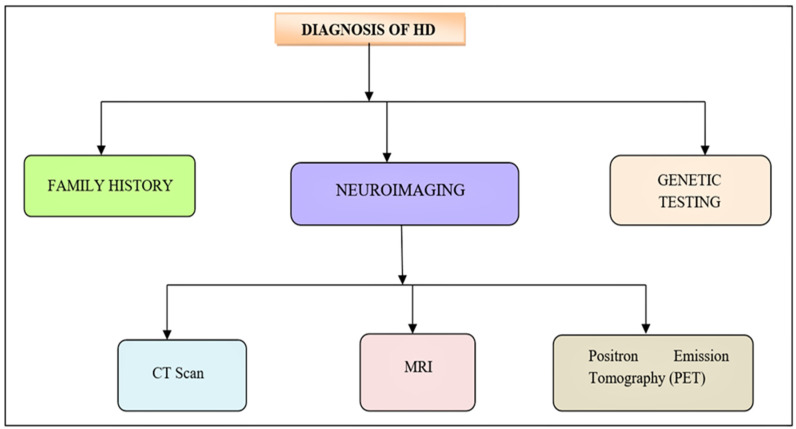
Background: Huntington's disease is an inherited autosomal dominant trait neuro-degenerative disorder caused by changes (mutations) of a gene called huntingtin (htt) that is located on the short arm (p) of chromosome 4, CAG expansion mutation. It is characterized by unusual movements, cognitive and psychiatric disorders.
Objective: This review was undertaken to apprehend biological pathways of Huntington's disease (HD) pathogenesis and its management by nature-derived products. Natural products can be lucrative for the management of HD as it shows protection against HD in pre-clinical trials. Advanced research is still required to assess the therapeutic effectiveness of the known organic products and their isolated compounds in HD experimental models.
Summary: Degeneration of neurons in Huntington's disease is distinguished by progressive loss of motor coordination and muscle function. This is due to the expansion of CAG trinucleotide in the first exon of the htt gene responsible for neuronal death and neuronal network degeneration in the brain. It is believed that the factors such as molecular genetics, oxidative stress, excitotoxicity, mitochondrial dysfunction, neuroglia dysfunction, protein aggregation, and altered UPS leads to HD. The defensive effect of the natural product provides therapeutic efficacy against HD. Recent reports on natural drugs have enlightened the protective role against HD via antioxidant, anti-inflammatory, antiapoptotic, and neurofunctional regulation.
Keywords: CAG expansion; Huntington’s disease (HD); huntingtin (htt); natural drugs; natural products; neurodegenerative disorder; pathogenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Bruyn G. Handbook of Clinical Neurology. Volume 4 Elsevier; Amsterdam, The Netherlands: 1968. (I0 Leucodystrophies Lipidoses).
Publication types
LinkOut - more resources
Full Text Sources
