

The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program
- PMID: 36291736
- PMCID: PMC9599222
- DOI: 10.3390/biom12101527
The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program
Abstract
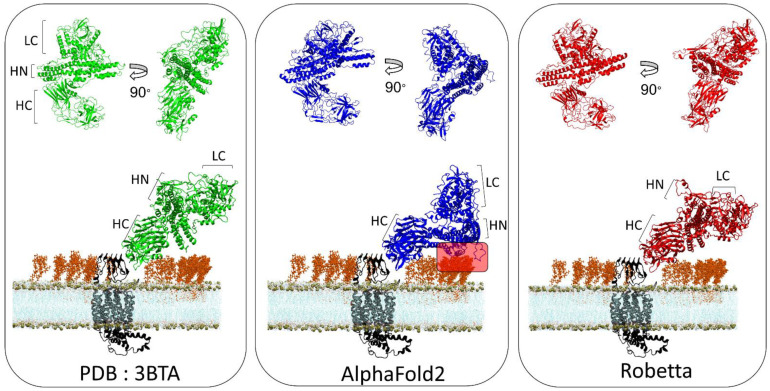
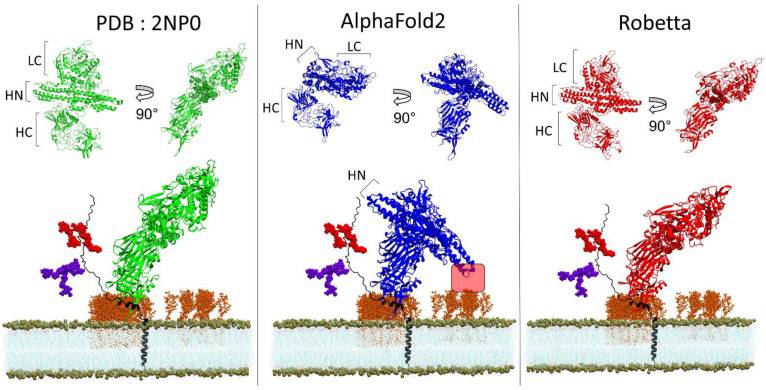
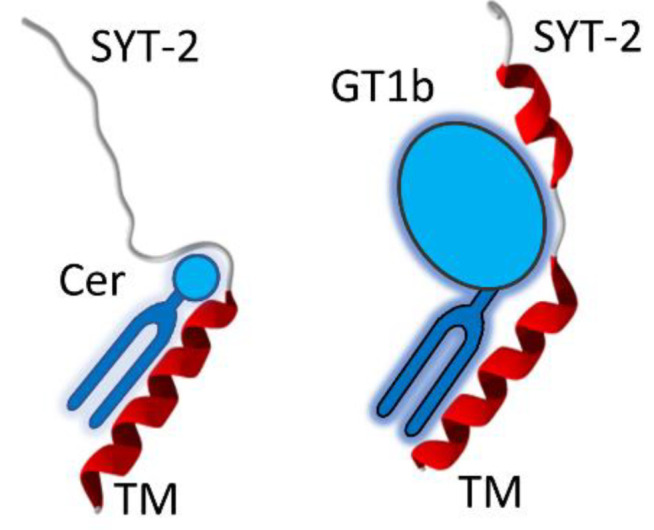
One of the most important lessons we have learned from sequencing the human genome is that not all proteins have a 3D structure. In fact, a large part of the human proteome is made up of intrinsically disordered proteins (IDPs) which can adopt multiple structures, and therefore, multiple functions, depending on the ligands with which they interact. Under these conditions, one can wonder about the value of algorithms developed for predicting the structure of proteins, in particular AlphaFold, an AI which claims to have solved the problem of protein structure. In a recent study, we highlighted a particular weakness of AlphaFold for membrane proteins. Based on this observation, we have proposed a paradigm, referred to as "Epigenetic Dimension of Protein Structure" (EDPS), which takes into account all environmental parameters that control the structure of a protein beyond the amino acid sequence (hence "epigenetic"). In this new study, we compare the reliability of the AlphaFold and Robetta algorithms' predictions for a new set of membrane proteins involved in human pathologies. We found that Robetta was generally more accurate than AlphaFold for ascribing a membrane-compatible topology. Raft lipids (e.g., gangliosides), which control the structural dynamics of membrane protein structure through chaperone effects, were identified as major actors of the EDPS paradigm. We conclude that the epigenetic dimension of a protein structure is an intrinsic weakness of AI-based protein structure prediction, especially AlphaFold, which warrants further development.
Keywords: AI; alphafold; ganglioside; lipid rafts; membrane; molecular modeling; pathology; protein structure; therapy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
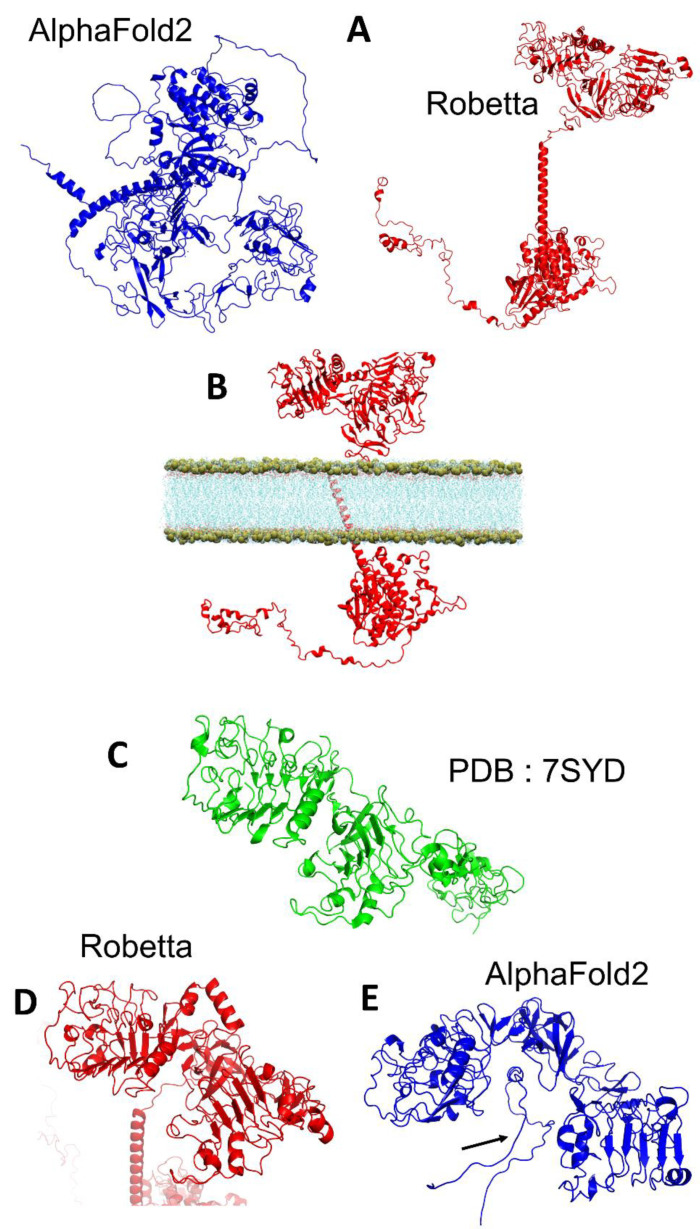
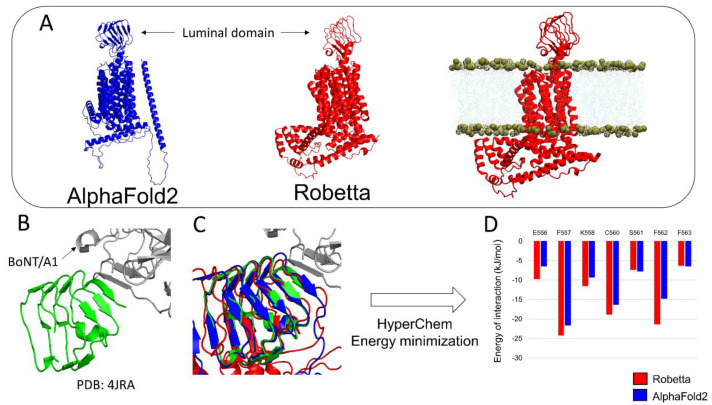
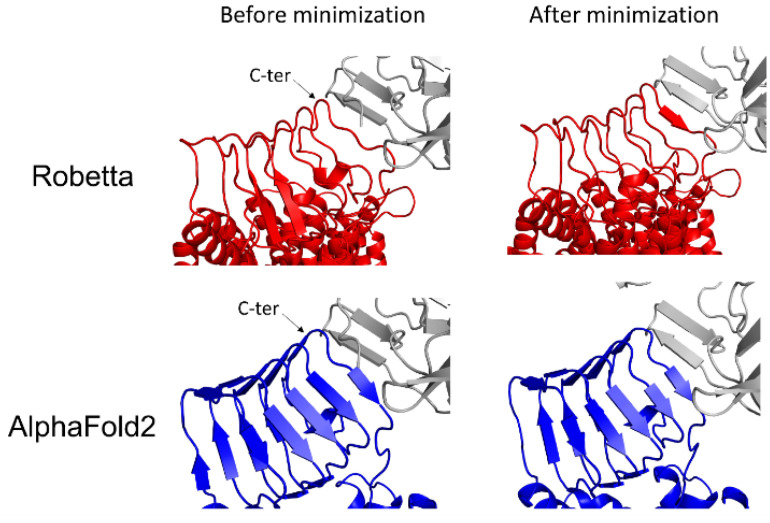
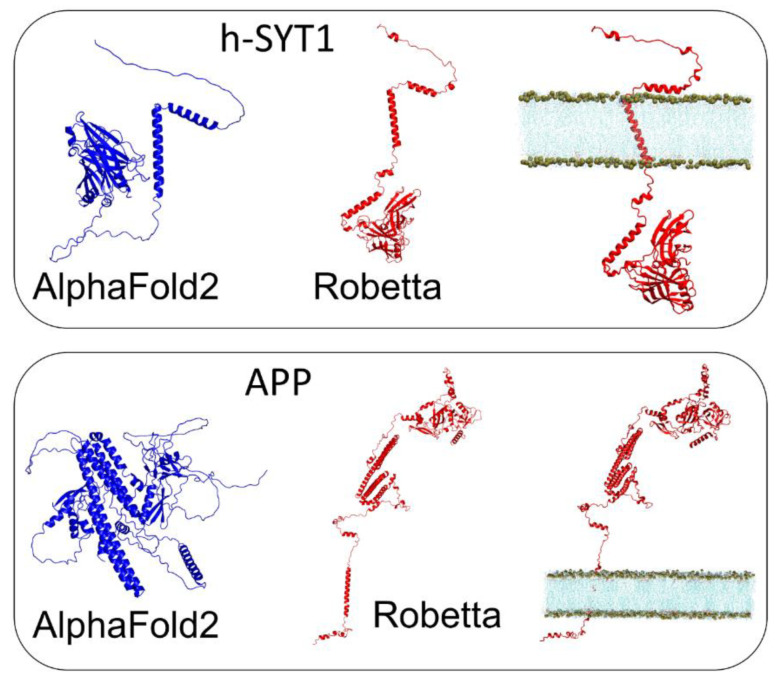

Similar articles
-
The epigenetic dimension of protein structure.Biomol Concepts. 2022 Feb 21;13(1):55-60. doi: 10.1515/bmc-2022-0006. Biomol Concepts. 2022. PMID: 35189052
-
AlphaFold and Implications for Intrinsically Disordered Proteins.J Mol Biol. 2021 Oct 1;433(20):167208. doi: 10.1016/j.jmb.2021.167208. Epub 2021 Aug 18. J Mol Biol. 2021. PMID: 34418423 Review.
-
Conformationally adaptive therapeutic peptides for diseases caused by intrinsically disordered proteins (IDPs). New paradigm for drug discovery: Target the target, not the arrow.Pharmacol Ther. 2025 Mar;267:108797. doi: 10.1016/j.pharmthera.2025.108797. Epub 2025 Jan 17. Pharmacol Ther. 2025. PMID: 39828029 Review.
-
Energy Landscapes of Protein Aggregation and Conformation Switching in Intrinsically Disordered Proteins.J Mol Biol. 2021 Oct 1;433(20):167182. doi: 10.1016/j.jmb.2021.167182. Epub 2021 Aug 3. J Mol Biol. 2021. PMID: 34358545 Review.
-
AlphaFold, Artificial Intelligence (AI), and Allostery.J Phys Chem B. 2022 Sep 1;126(34):6372-6383. doi: 10.1021/acs.jpcb.2c04346. Epub 2022 Aug 17. J Phys Chem B. 2022. PMID: 35976160 Free PMC article. Review.
Cited by
-
Advancing membrane-associated protein docking with improved sampling and scoring in Rosetta.bioRxiv [Preprint]. 2024 Jul 13:2024.07.09.602802. doi: 10.1101/2024.07.09.602802. bioRxiv. 2024. Update in: J Chem Theory Comput. 2024 Dec 10;20(23):10740-10749. doi: 10.1021/acs.jctc.4c00927. PMID: 39026849 Free PMC article. Updated. Preprint.
-
Advancing Membrane-Associated Protein Docking with Improved Sampling and Scoring in Rosetta.J Chem Theory Comput. 2024 Dec 10;20(23):10740-10749. doi: 10.1021/acs.jctc.4c00927. Epub 2024 Nov 22. J Chem Theory Comput. 2024. PMID: 39574325
-
Predictive Modeling of Proteins Encoded by a Plant Virus Sheds a New Light on Their Structure and Inherent Multifunctionality.Biomolecules. 2024 Jan 2;14(1):62. doi: 10.3390/biom14010062. Biomolecules. 2024. PMID: 38254661 Free PMC article.
-
Structural Basis of Botulinum Toxin Type F Binding to Glycosylated Human SV2A: In Silico Studies at the Periphery of a Lipid Raft.Biomolecules. 2022 Dec 6;12(12):1821. doi: 10.3390/biom12121821. Biomolecules. 2022. PMID: 36551250 Free PMC article.
-
Inferring molecular inhibition potency with AlphaFold predicted structures.Sci Rep. 2024 Apr 8;14(1):8252. doi: 10.1038/s41598-024-58394-z. Sci Rep. 2024. PMID: 38589418 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
