

Targeted Anti-Mitochondrial Therapy: The Future of Oncology
- PMID: 36292613
- PMCID: PMC9602426
- DOI: 10.3390/genes13101728
Targeted Anti-Mitochondrial Therapy: The Future of Oncology
Abstract
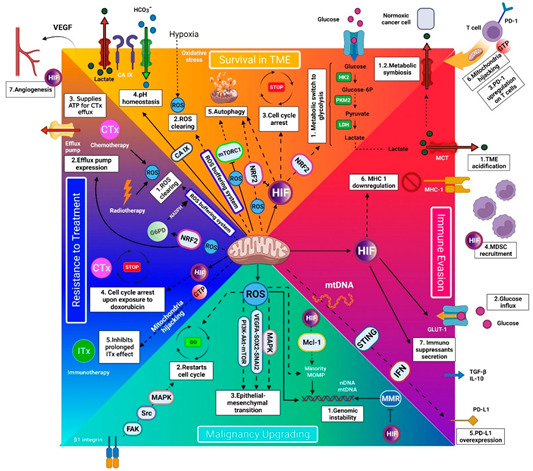
Like living organisms, cancer cells require energy to survive and interact with their environment. Mitochondria are the main organelles for energy production and cellular metabolism. Recently, investigators demonstrated that cancer cells can hijack mitochondria from immune cells. This behavior sheds light on a pivotal piece in the cancer puzzle, the dependence on the normal cells. This article illustrates the benefits of new functional mitochondria for cancer cells that urge them to hijack mitochondria. It describes how functional mitochondria help cancer cells' survival in the harsh tumor microenvironment, immune evasion, progression, and treatment resistance. Recent evidence has put forward the pivotal role of mitochondria in the metabolism of cancer stem cells (CSCs), the tumor components responsible for cancer recurrence and metastasis. This theory highlights the mitochondria in cancer biology and explains how targeting mitochondria may improve oncological outcomes.
Keywords: ATP; T cell; cancer cell; cancer stem cell; cancer treatment; mitochondria.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Halvorson H.M., Wyatt K.H., Kuehn K.A. Ecological significance of autotroph–heterotroph microbial interactions in freshwaters. Freshw. Biol. 2020;65:1183–1188. doi: 10.1111/fwb.13530. - DOI
-
- Akbari H., Taghizadeh-Hesary F., Heike Y., Bahadori M. Cell Energy: A New Hypothesis in Decoding Cancer Evolution. Arch. Iran. Med. 2019;22:733–735. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
