

Correlation between Phenotype and Genotype in CTNNB1 Syndrome: A Systematic Review of the Literature
- PMID: 36293418
- PMCID: PMC9604177
- DOI: 10.3390/ijms232012564
Correlation between Phenotype and Genotype in CTNNB1 Syndrome: A Systematic Review of the Literature
Abstract
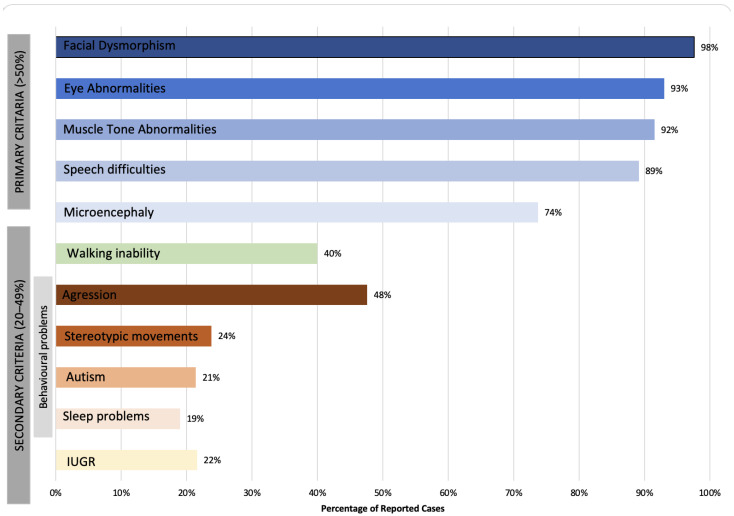
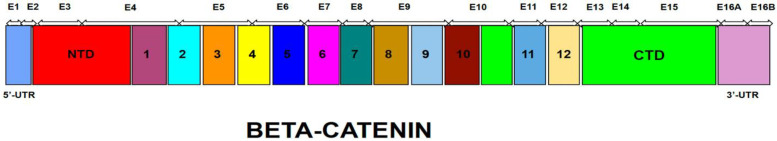
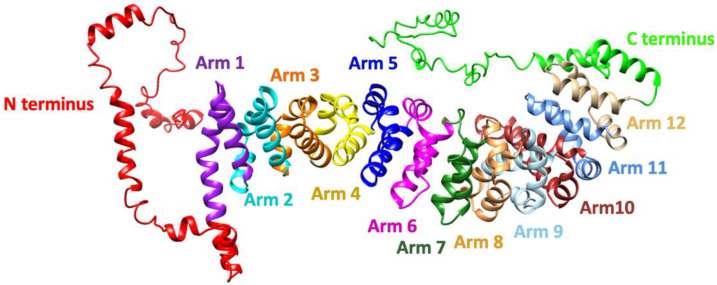
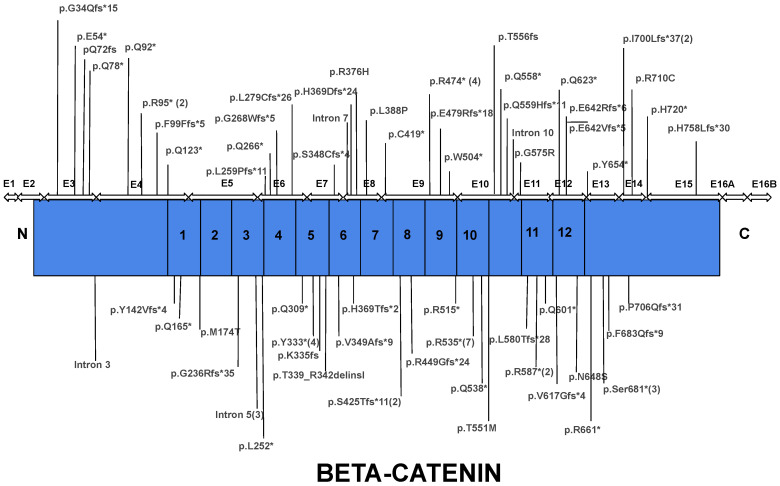
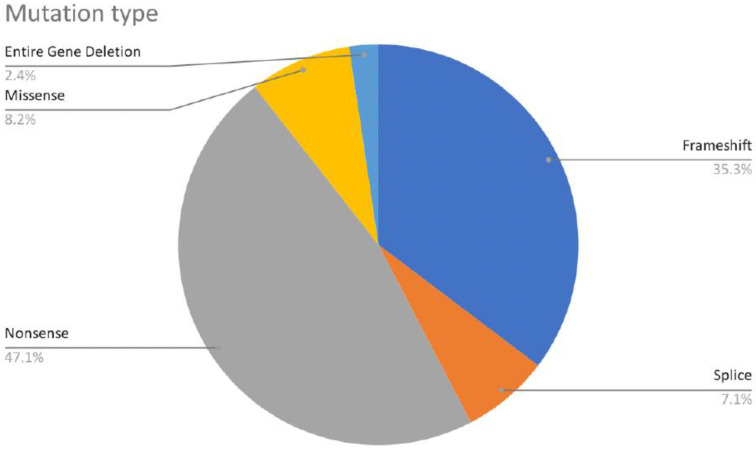
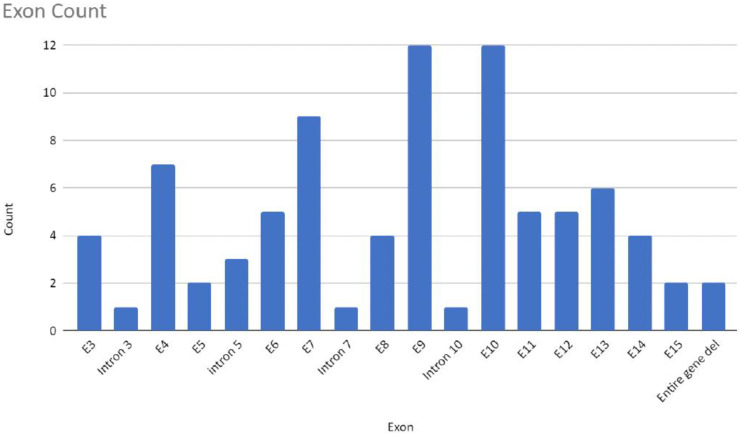
The CTNNB1 Syndrome is a rare neurodevelopmental disorder associated with developmental delay, intellectual disability, and delayed or absent speech. The aim of the present study is to systematically review the available data on the prevalence of clinical manifestations and to evaluate the correlation between phenotype and genotype in published cases of patients with CTNNB1 Syndrome. Studies were identified by systematic searches of four major databases. Information was collected on patients' genetic mutations, prenatal and neonatal problems, head circumference, muscle tone, EEG and MRI results, dysmorphic features, eye abnormalities, early development, language and comprehension, behavioral characteristics, and additional clinical problems. In addition, the mutations were classified into five groups according to the severity of symptoms. The study showed wide genotypic and phenotypic variability in patients with CTNNB1 Syndrome. The most common moderate-severe phenotype manifested in facial dysmorphisms, microcephaly, various motor disabilities, language and cognitive impairments, and behavioral abnormalities (e.g., autistic-like or aggressive behavior). Nonsense and missense mutations occurring in exons 14 and 15 were classified in the normal clinical outcome category/group because they had presented an otherwise normal phenotype, except for eye abnormalities. A milder phenotype was also observed with missense and nonsense mutations in exon 13. The autosomal dominant CTNNB1 Syndrome encompasses a wide spectrum of clinical features, ranging from normal to severe. While mutations cannot be more generally categorized by location, it is generally observed that the C-terminal protein region (exons 13, 14, 15) correlates with a milder phenotype.
Keywords: beta-catenin; eye movement disorders; hypotonia; intellectual disability; loss of function mutation; microcephaly.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Verhoeven W.M.A., Egger J.I.M., Jongbloed R.E., van Putten M.M., de Bruin-van Zandwijk M., Zwemer A.S., Pfundt R., Willemsen M.H. A de novo CTNNB1 Novel Splice Variant in an Adult Female with Severe Intellectual Disability. Int. Med. Case Rep. J. 2020;13:487–492. doi: 10.2147/IMCRJ.S270487. - DOI - PMC - PubMed
-
- Kuechler A., Willemsen M.H., Albrecht B., Bacino C.A., Bartholomew D.W., van Bokhoven H., van den Boogaard M.J., Bramswig N., Buttner C., Cremer K., et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: Expanding the mutational and clinical spectrum. Hum. Genet. 2015;134:97–109. doi: 10.1007/s00439-014-1498-1. - DOI - PubMed
-
- de Ligt J., Willemsen M.H., van Bon B.W., Kleefstra T., Yntema H.G., Kroes T., Vulto-van Silfhout A.T., Koolen D.A., de Vries P., Gilissen C., et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
