

Advances in Fungal Phenaloenones-Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors
- PMID: 36296388
- PMCID: PMC9608606
- DOI: 10.3390/molecules27206797
Advances in Fungal Phenaloenones-Natural Metabolites with Great Promise: Biosynthesis, Bioactivities, and an In Silico Evaluation of Their Potential as Human Glucose Transporter 1 Inhibitors
Abstract
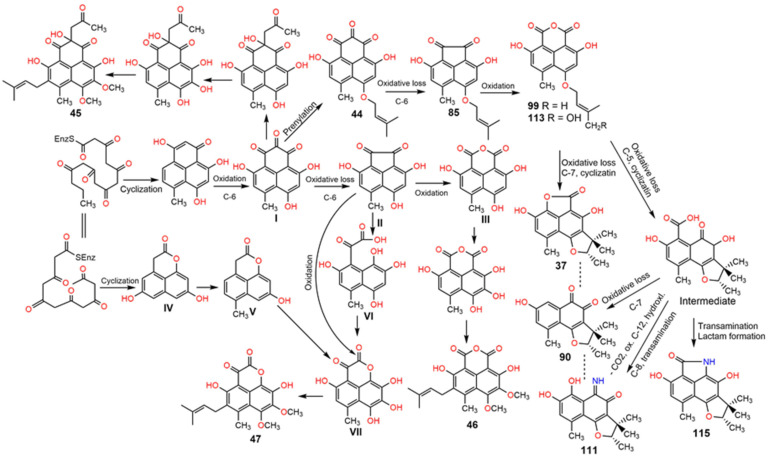
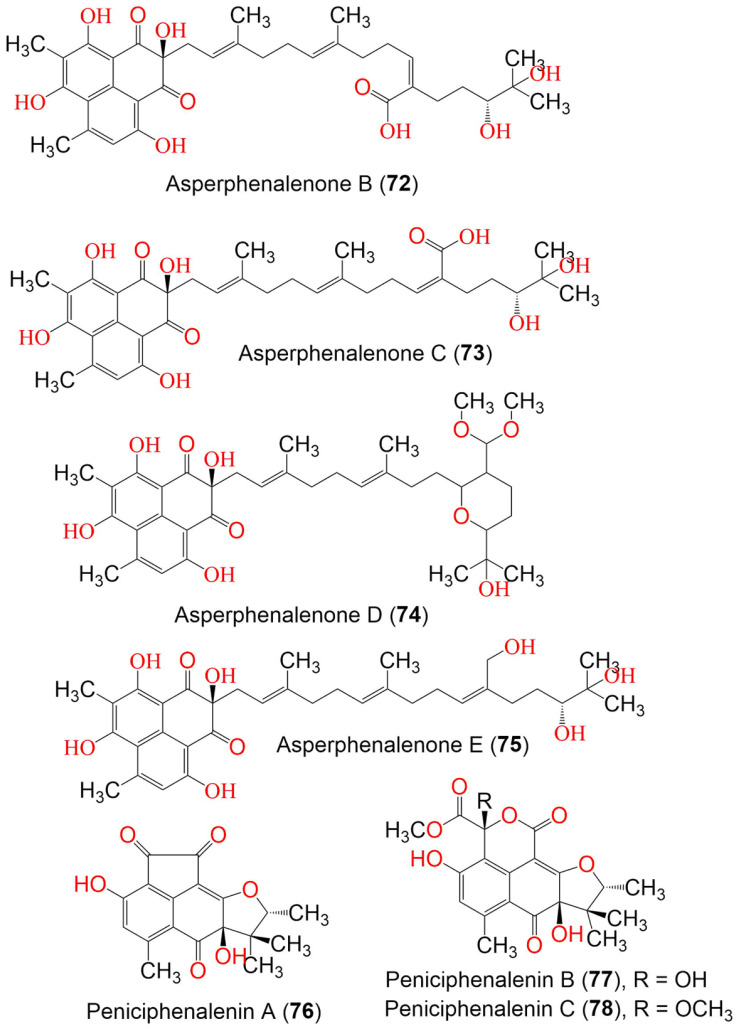
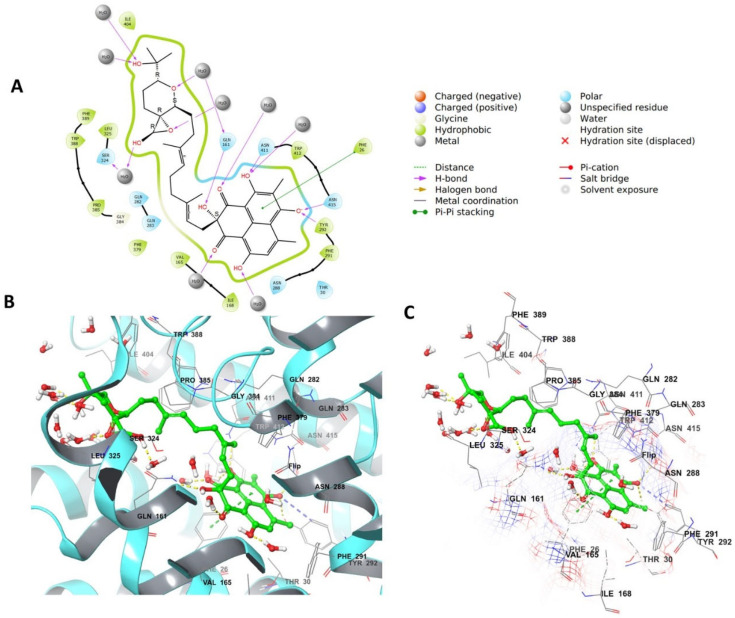
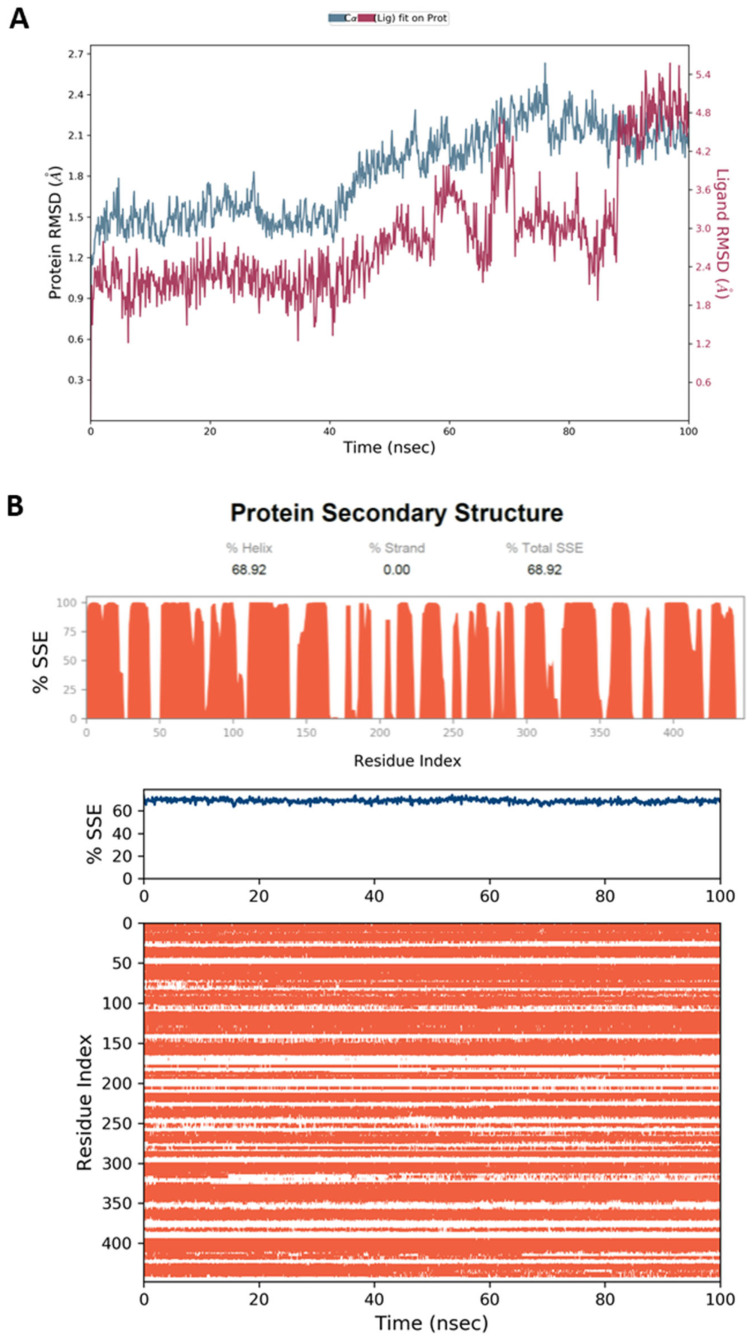
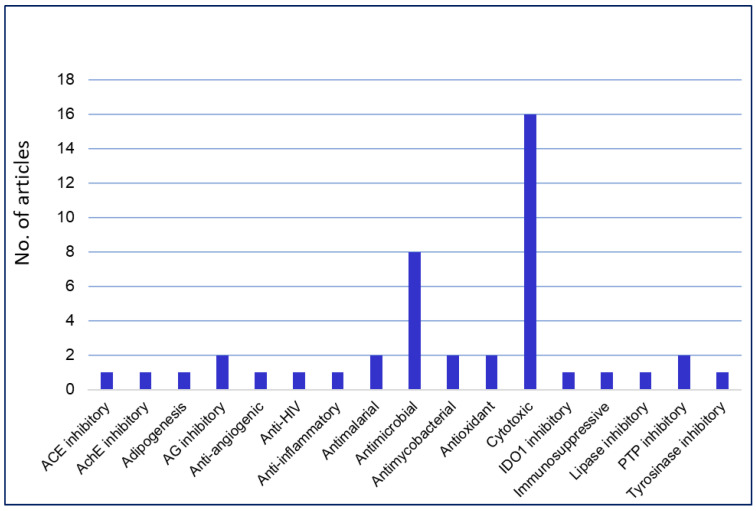
Phenaloenones are structurally unique aromatic polyketides that have been reported in both microbial and plant sources. They possess a hydroxy perinaphthenone three-fused-ring system and exhibit diverse bioactivities, such as cytotoxic, antimicrobial, antioxidant, and anti-HIV properties, and tyrosinase, α-glucosidase, lipase, AchE (acetylcholinesterase), indoleamine 2,3-dioxygenase 1, angiotensin-I-converting enzyme, and tyrosine phosphatase inhibition. Moreover, they have a rich nucleophilic nucleus that has inspired many chemists and biologists to synthesize more of these related derivatives. The current review provides an overview of the reported phenalenones with a fungal origin, including their structures, sources, biosynthesis, and bioactivities. Moreover, more than 135 metabolites have been listed, and 71 references have been cited. SuperPred, an artificial intelligence (AI) webserver, was used to predict the potential targets for selected phenalenones. Among these targets, we chose human glucose transporter 1 (hGLUT1) for an extensive in silico study, as it shows high probability and model accuracy. Among them, aspergillussanones C (60) and G (60) possessed the highest negative docking scores of -15.082 and -14.829 kcal/mol, respectively, compared to the native inhibitor of 5RE (score: -11.206 kcal/mol). The MD (molecular dynamics) simulation revealed their stability in complexes with GLUT1 at 100 ns. The virtual screening study results open up a new therapeutic approach by using some phenalenones as hGLUT1 inhibitors, which might be a potential target for cancer therapy.
Keywords: bioactivities; biosynthesis; fungi; human glucose transporter 1 (hGLUT1) inhibitors; in silico screening; molecular docking; molecular dynamics; phenaloenones.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

























References
-
- Ibrahim S.R.M., Mohamed S.G.A., Sindi I.A., Mohamed G.A. Biologically active secondary metabolites and biotechnological applications of species of the family Chaetomiaceae (Sordariales): An updated review from 2016 to 2021. Mycol. Prog. 2021;20:595–639. doi: 10.1007/s11557-021-01704-w. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
