

Role of non-cardiomyocytes in anticancer drug-induced cardiotoxicity: A systematic review
- PMID: 36300001
- PMCID: PMC9589207
- DOI: 10.1016/j.isci.2022.105283
Role of non-cardiomyocytes in anticancer drug-induced cardiotoxicity: A systematic review
Abstract
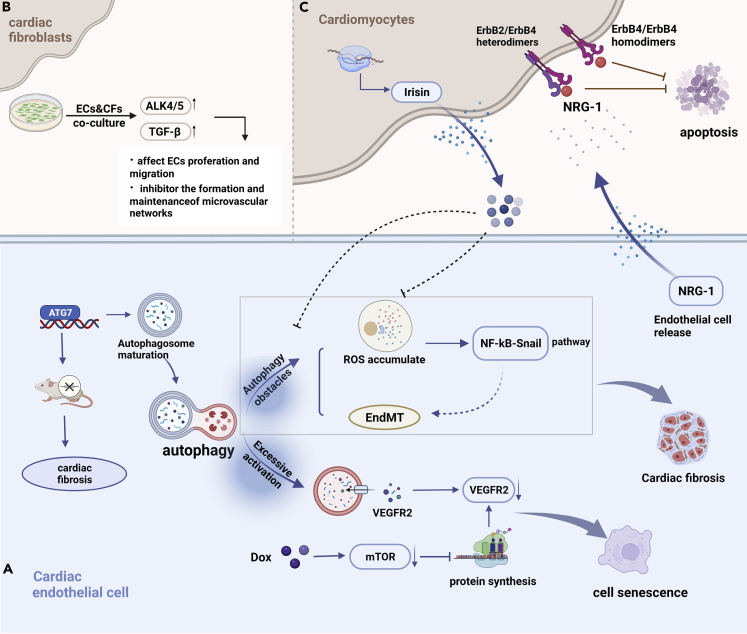
Cardiotoxicity induced by anticancer drugs interferes with the continuation of optimal treatment, inducing life-threatening risks or leading to long-term morbidity. The heart is a complex pluricellular organ comprised of cardiomyocytes and non-cardiomyocytes. Although the study of these cell populations has been often focusing on cardiomyocytes, the contributions of non-cardiomyocytes to development and disease are increasingly being appreciated as both dynamic and essential. This review summarized the role of non-cardiomyocytes in anticancer drug-induced cardiotoxicity, including the mechanism of direct damage to resident non-cardiomyocytes, cardiomyocytes injury caused by paracrine modality, myocardial inflammation induced by transient cell populations and the protective agents that focused on non-cardiomyocytes.
Keywords: Cardiovascular medicine; cancer; toxicity assessment.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- Ahmed S.H., Clark L.L., Pennington W.R., Webb C.S., Bonnema D.D., Leonardi A.H., Mcclure C.D., Spinale F.G., Zile M.R. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–2096. - PubMed
-
- Ando J., Yamamoto K. Flow detection and calcium signalling in vascular endothelial cells. Cardiovasc. Res. 2013;99:260–268. - PubMed
-
- Banerjee I., Fuseler J.W., Price R.L., Borg T.K., Baudino T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H1883–H1891. - PubMed
-
- Brutsaert D.L. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003;83:59–115. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
