

Multireference Generalization of the Weighted Thermodynamic Perturbation Method
- PMID: 36301936
- PMCID: PMC9771595
- DOI: 10.1021/acs.jpca.2c06201
Multireference Generalization of the Weighted Thermodynamic Perturbation Method
Abstract
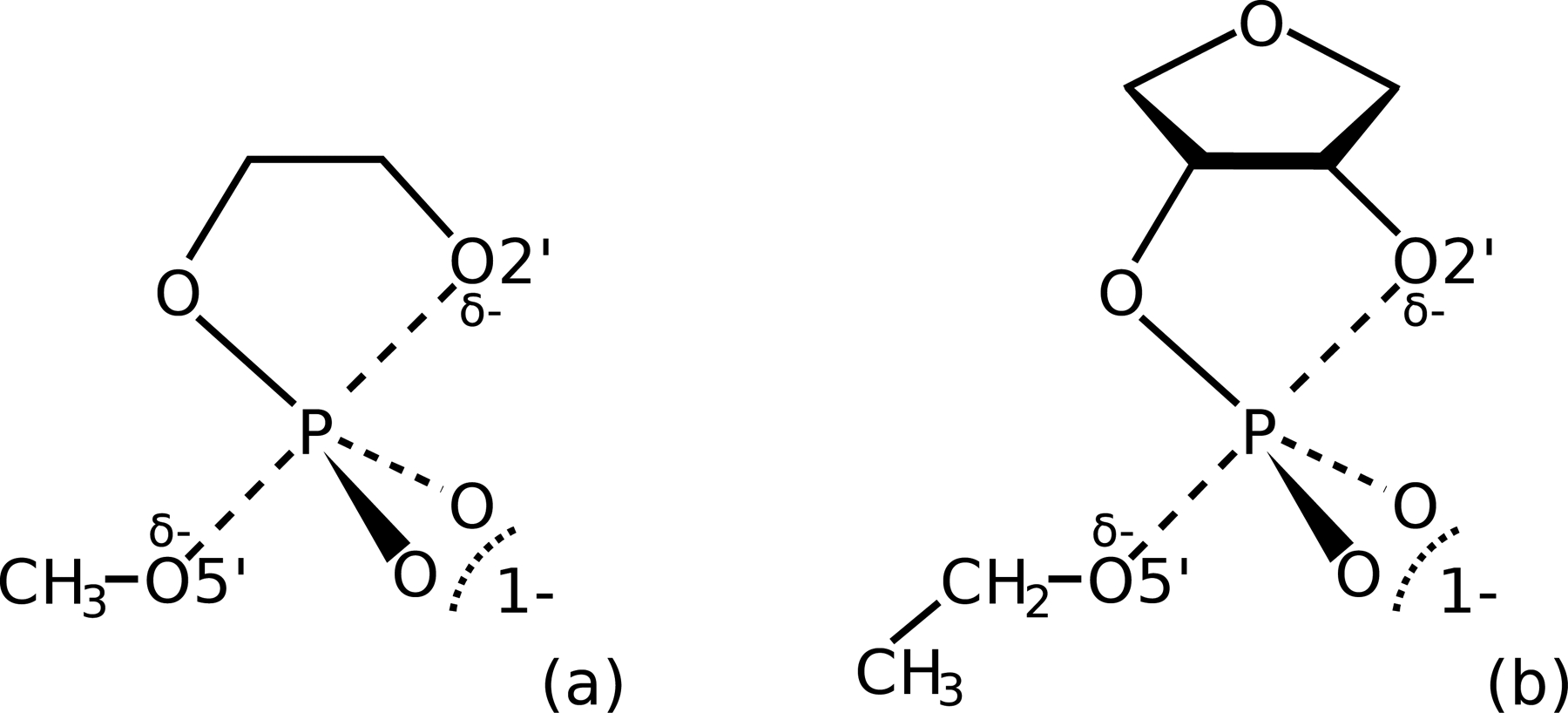
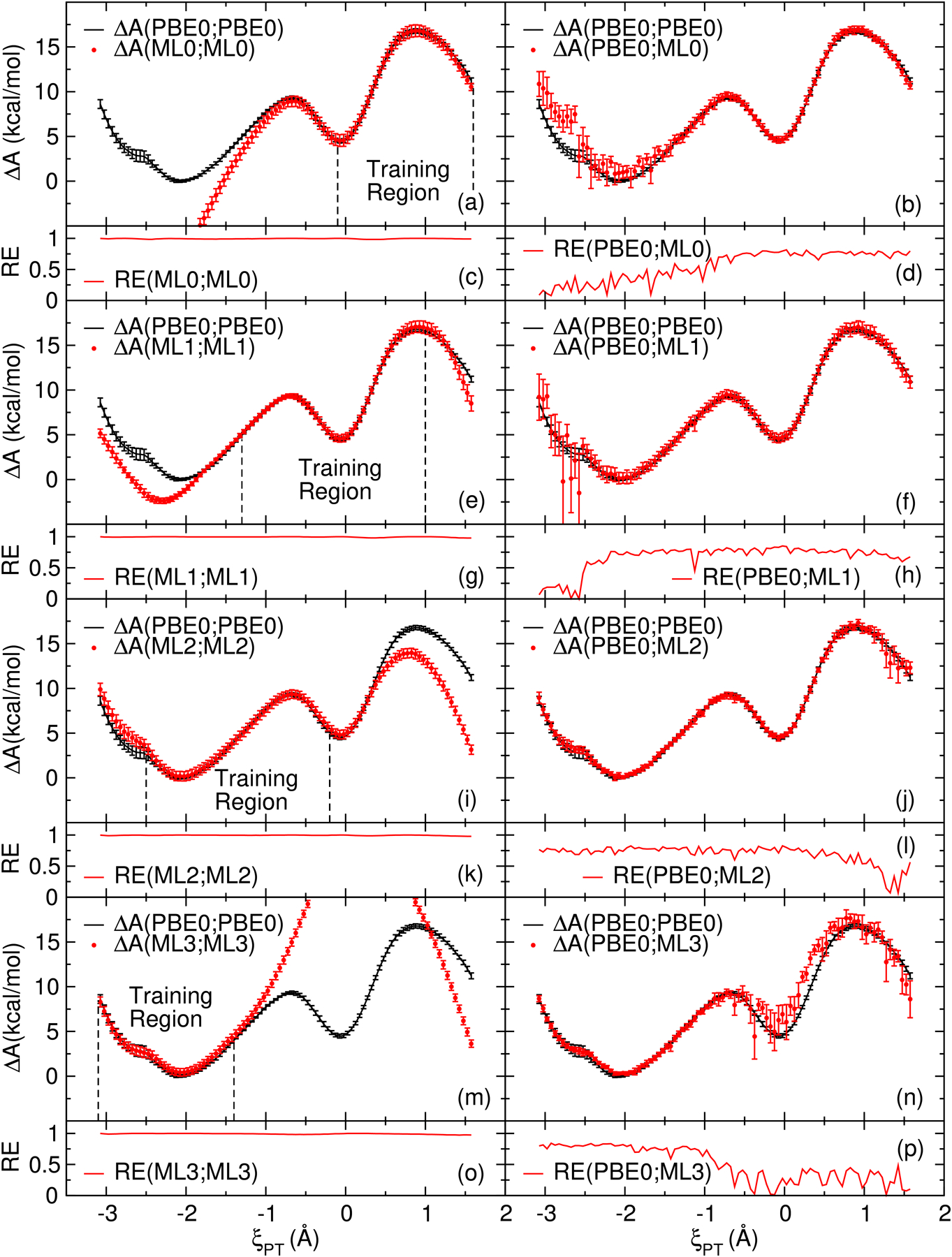
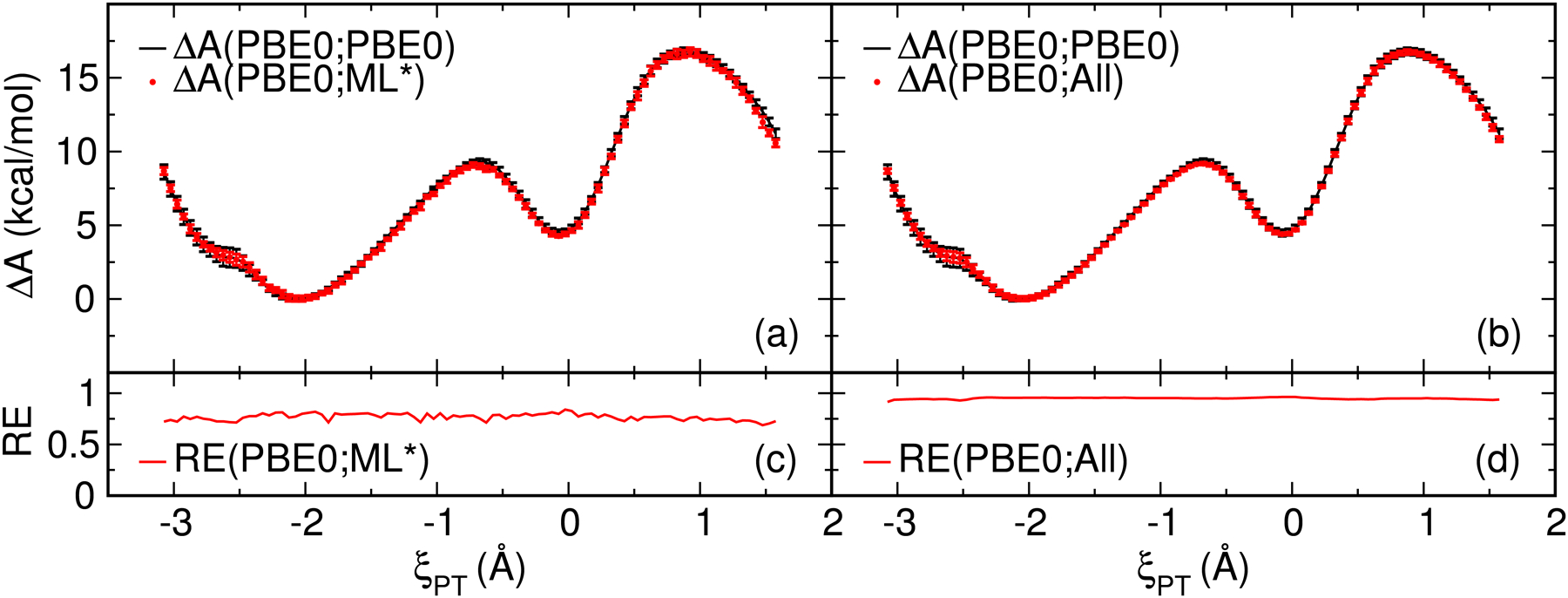
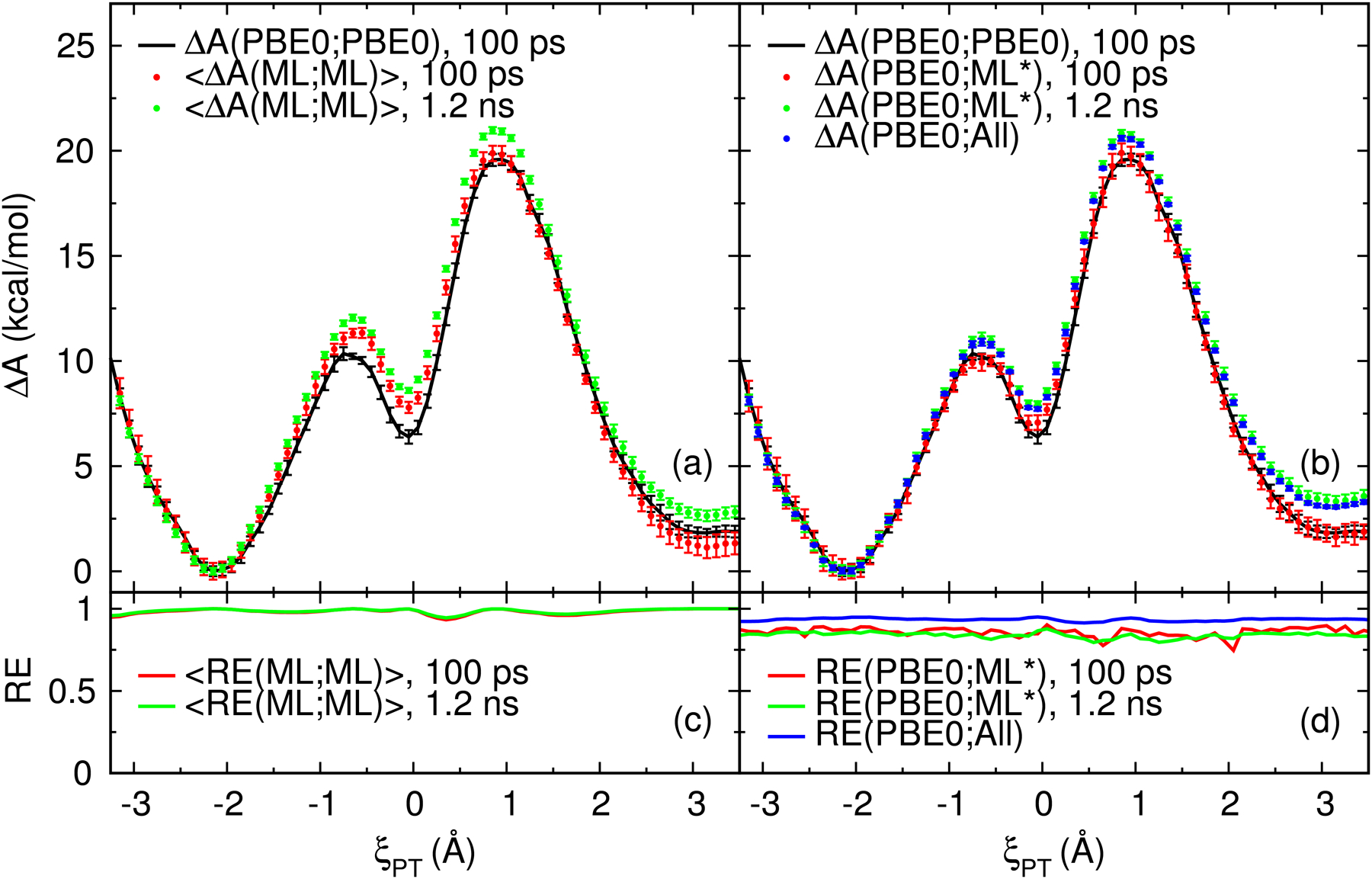
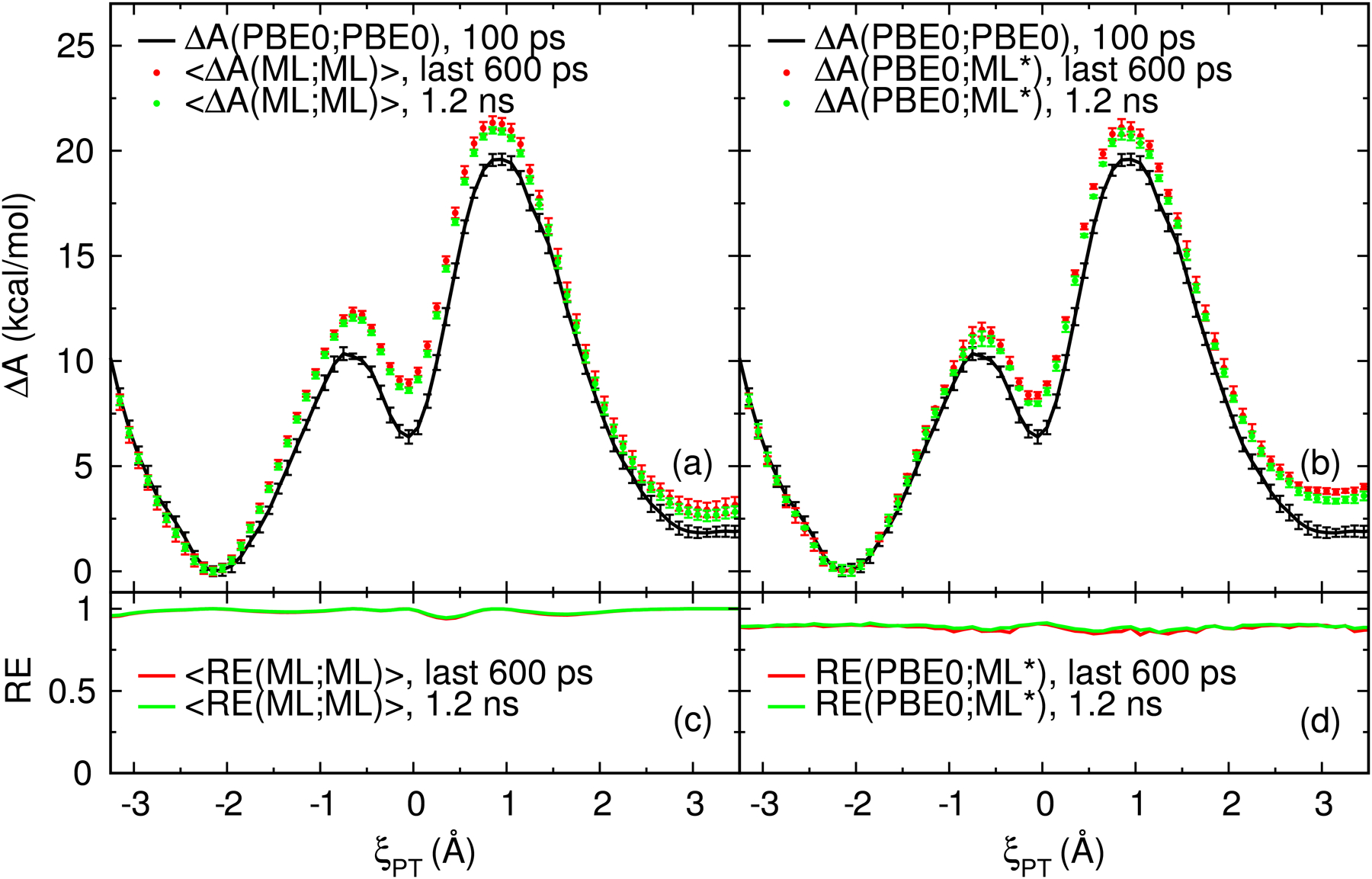
We describe the generalized weighted thermodynamic perturbation (gwTP) method for estimating the free energy surface of an expensive "high-level" potential energy function from the umbrella sampling performed with multiple inexpensive "low-level" reference potentials. The gwTP method is a generalization of the weighted thermodynamic perturbation (wTP) method developed by Li and co-workers [J. Chem. Theory Comput. 2018, 14, 5583-5596] that uses a single "low-level" reference potential. The gwTP method offers new possibilities in model design whereby the sampling generated from several low-level potentials may be combined (e.g., specific reaction parameter models that might have variable accuracy at different stages of a multistep reaction). The gwTP method is especially well suited for use with machine learning potentials (MLPs) that are trained against computationally expensive ab initio quantum mechanical/molecular mechanical (QM/MM) energies and forces using active learning procedures that naturally produce multiple distinct neural network potentials. Simulations can be performed with greater sampling using the fast MLPs and then corrected to the ab initio level using gwTP. The capabilities of the gwTP method are demonstrated by creating reference potentials based on the MNDO/d and DFTB2/MIO semiempirical models supplemented with the "range-corrected deep potential" (DPRc). The DPRc parameters are trained to ab initio QM/MM data, and the potentials are used to calculate the free energy surface of stepwise mechanisms for nonenzymatic RNA 2'-O-transesterification model reactions. The extended sampling made possible by the reference potentials allows one to identify unequilibrated portions of the simulations that are not always evident from the short time scale commonly used with ab initio QM/MM potentials. We show that the reference potential approach can yield more accurate ab initio free energy predictions than the wTP method or what can be reasonably afforded from explicit ab initio QM/MM sampling.
Figures
References
-
- Klippenstein SJ; Pande VS; Truhlar DG Chemical Kinetics and Mechanisms of Complex Systems: A Perspective on Recent Theoretical Advances. J. Am. Chem. Soc 2014, 136, 528–46. - PubMed
-
- Wüthrich K, Grubbs RH, de Bocarmé TV, De Wit A, Eds. Catalysis in Chemistry and Biology; World Scientific, 2018.
-
- Cheng G-J; Zhang X; Wa Chung L; Xu L; Wu Y-D Computational Organic Chemistry: Bridging Theory and Experiment in Establishing the Mechanisms of Chemical Reactions. J. Am. Chem. Soc 2015, 137, 1706–1725. - PubMed
-
- Talanquer V Importance of Understanding Fundamental Chemical Mechanisms. J. Chem. Educ 2018, 95, 1905–1911.
-
- Christ C; Mark A; van Gunsteren W Basic Ingredients of Free Energy Calculations: A Review. J. Comput. Chem 2010, 31, 1569–1582. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
