

Genome sequence of the entomopathogenic Serratia entomophila isolate 626 and characterisation of the species specific itaconate degradation pathway
- PMID: 36303123
- PMCID: PMC9615192
- DOI: 10.1186/s12864-022-08938-2
Genome sequence of the entomopathogenic Serratia entomophila isolate 626 and characterisation of the species specific itaconate degradation pathway
Abstract
Background: Isolates of Serratia entomophila and S. proteamaculans (Yersiniaceae) cause disease specific to the endemic New Zealand pasture pest, Costelytra giveni (Coleoptera: Scarabaeidae). Previous genomic profiling has shown that S. entomophila isolates appear to have conserved genomes and, where present, conserved plasmids. In the absence of C. giveni larvae, S. entomophila prevalence reduces in the soil over time, suggesting that S. entomophila has formed a host-specific relationship with C. giveni. To help define potential genetic mechanisms driving retention of the chronic disease of S. entomophila, the genome of the isolate 626 was sequenced, enabling the identification of unique chromosomal properties, and defining the gain/loss of accessory virulence factors relevant to pathogenicity to C. giveni larvae.
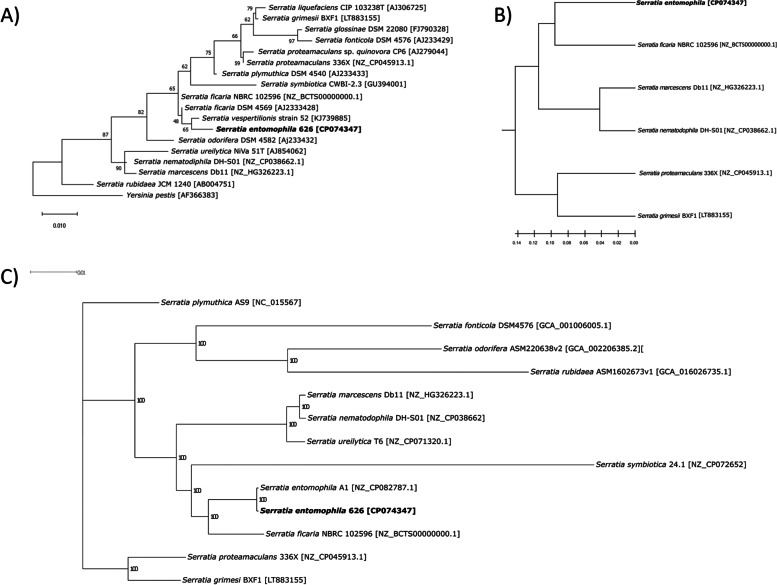
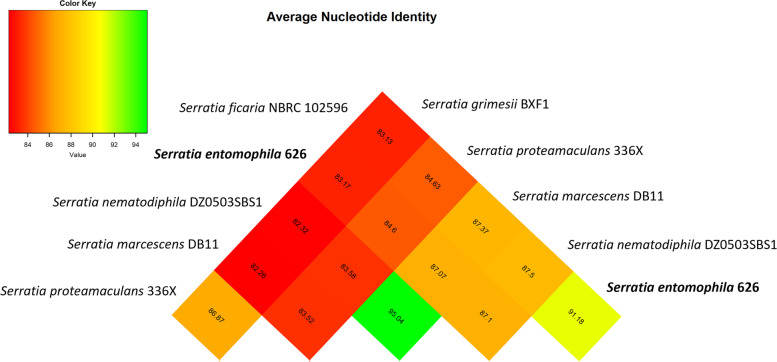
Results: We report the complete sequence of S. entomophila isolate 626, a causal agent of amber disease in C. giveni larvae. The genome of S. entomophila 626 is 5,046,461 bp, with 59.1% G + C content and encoding 4,695 predicted CDS. Comparative analysis with five previously sequenced Serratia species, S. proteamaculans 336X, S. marcescens Db11, S. nematodiphila DH-S01, S. grimesii BXF1, and S. ficaria NBRC 102596, revealed a core of 1,165 genes shared. Further comparisons between S. entomophila 626 and S. proteamaculans 336X revealed fewer predicted phage-like regions and genomic islands in 626, suggesting less horizontally acquired genetic material. Genomic analyses revealed the presence of a four-gene itaconate operon, sharing a similar gene order as the Yersinia pestis ripABC complex. Assessment of a constructed 626::RipC mutant revealed that the operon confer a possible metabolic advantage to S. entomophila in the initial stages of C. giveni infection.
Conclusions: Evidence is presented where, relative to S. proteamaculans 336X, S. entomophila 626 encodes fewer genomic islands and phages, alluding to limited horizontal gene transfer in S. entomophila. Bioassay assessments of a S. entomophila-mutant with a targeted mutation of the itaconate degradation region unique to this species, found the mutant to have a reduced capacity to replicate post challenge of the C. giveni larval host, implicating the itaconate operon in establishment within the host.
Keywords: Chromosome; Entomopathogen; Genome; Horizontal gene transfer; Itaconate; Virulence.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures










References
-
- Petersen LM, Tisa LS. Friend or foe? A review of the mechanisms that drive Serratia towards diverse lifestyles. Can J Microbiol. 2013;59(9):627–640. - PubMed
-
- Costa MAA, Owen RA, Tammsalu T, Buchanan G, Palmer T, Sargent F. Controlling and co-ordinating chitinase secretion in a Serratia marcescens population. Microbiology. 2019;165(11):1233–1244. - PubMed
-
- Jackson T. Biological control of grass grub in Canterbury. Proc NZ Grassl Assoc. 1990;52:217–20.
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
