

The multiple de novo copy number variant (MdnCNV) phenomenon presents with peri-zygotic DNA mutational signatures and multilocus pathogenic variation
- PMID: 36303224
- PMCID: PMC9609164
- DOI: 10.1186/s13073-022-01123-w
The multiple de novo copy number variant (MdnCNV) phenomenon presents with peri-zygotic DNA mutational signatures and multilocus pathogenic variation
Abstract
Background: The multiple de novo copy number variant (MdnCNV) phenotype is described by having four or more constitutional de novo CNVs (dnCNVs) arising independently throughout the human genome within one generation. It is a rare peri-zygotic mutational event, previously reported to be seen once in every 12,000 individuals referred for genome-wide chromosomal microarray analysis due to congenital abnormalities. These rare families provide a unique opportunity to understand the genetic factors of peri-zygotic genome instability and the impact of dnCNV on human diseases.
Methods: Chromosomal microarray analysis (CMA), array-based comparative genomic hybridization, short- and long-read genome sequencing (GS) were performed on the newly identified MdnCNV family to identify de novo mutations including dnCNVs, de novo single-nucleotide variants (dnSNVs), and indels. Short-read GS was performed on four previously published MdnCNV families for dnSNV analysis. Trio-based rare variant analysis was performed on the newly identified individual and four previously published MdnCNV families to identify potential genetic etiologies contributing to the peri-zygotic genomic instability. Lin semantic similarity scores informed quantitative human phenotype ontology analysis on three MdnCNV families to identify gene(s) driving or contributing to the clinical phenotype.
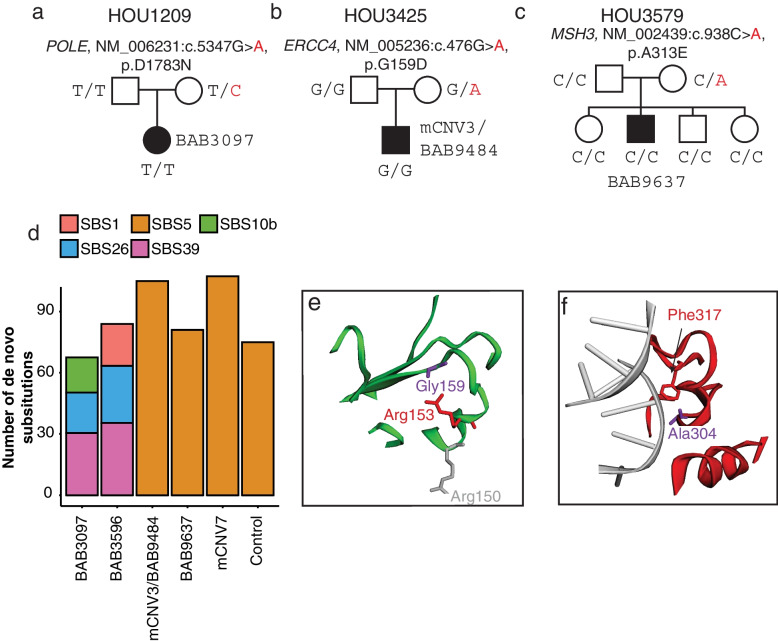
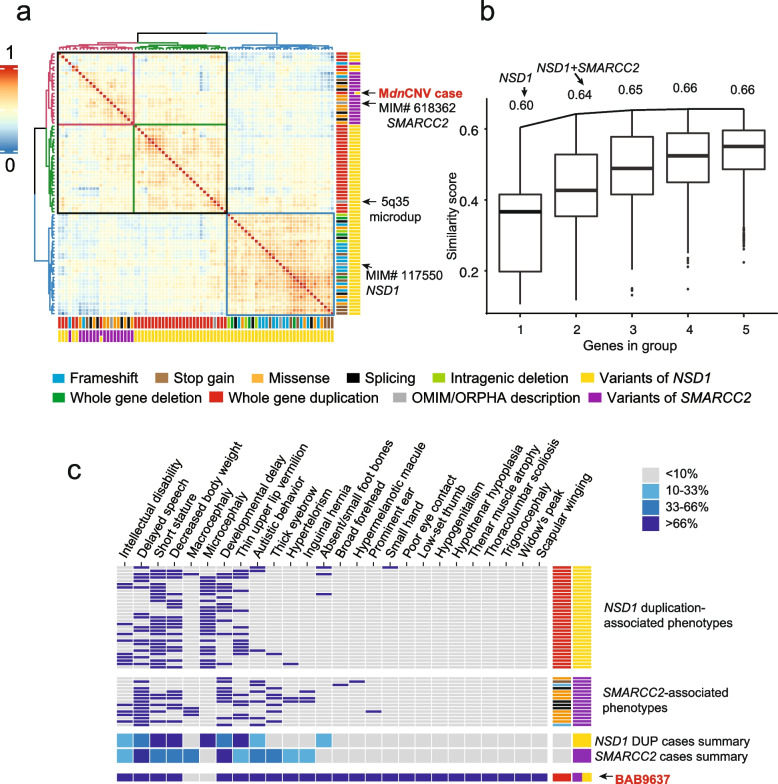
Results: In the newly identified MdnCNV case, we revealed eight de novo tandem duplications, each ~ 1 Mb, with microhomology at 6/8 breakpoint junctions. Enrichment of de novo single-nucleotide variants (SNV; 6/79) and de novo indels (1/12) was found within 4 Mb of the dnCNV genomic regions. An elevated post-zygotic SNV mutation rate was observed in MdnCNV families. Maternal rare variant analyses identified three genes in distinct families that may contribute to the MdnCNV phenomenon. Phenotype analysis suggests that gene(s) within dnCNV regions contribute to the observed proband phenotype in 3/3 cases. CNVs in two cases, a contiguous gene duplication encompassing PMP22 and RAI1 and another duplication affecting NSD1 and SMARCC2, contribute to the clinically observed phenotypic manifestations.
Conclusions: Characteristic features of dnCNVs reported here are consistent with a microhomology-mediated break-induced replication (MMBIR)-driven mechanism during the peri-zygotic period. Maternal genetic variants in DNA repair genes potentially contribute to peri-zygotic genomic instability. Variable phenotypic features were observed across a cohort of three MdnCNV probands, and computational quantitative phenotyping revealed that two out of three had evidence for the contribution of more than one genetic locus to the proband's phenotype supporting the hypothesis of de novo multilocus pathogenic variation (MPV) in those families.
Keywords: De novo CNV; De novo SNV, Human Phenotype Ontology, Structural variation; Genomic data integration, Genomic data visualization, MMBIR; Genomic instability; Long-read sequencing; Tandem duplication.
© 2022. The Author(s).
Conflict of interest statement
Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical chromosome microarray analysis (CMA) and other genomic studies (ES, genome sequencing) for patient/family care. J.R.L. serves on the Scientific Advisory Board of BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. PL and WB are employees of BCM and derive support through a professional service agreement with BG. MP, EH, and SJ are employees of Oxford Nanopore Technologies and are shareholders and/or share option holders. FJS has multiple travels sponsored by Pacbio and ONT. The remaining authors declare that they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
