

Computational Identification of Functional Centers in Complex Proteins: A Step-by-Step Guide With Examples
- PMID: 36303732
- PMCID: PMC9581015
- DOI: 10.3389/fbinf.2021.652286
Computational Identification of Functional Centers in Complex Proteins: A Step-by-Step Guide With Examples
Abstract
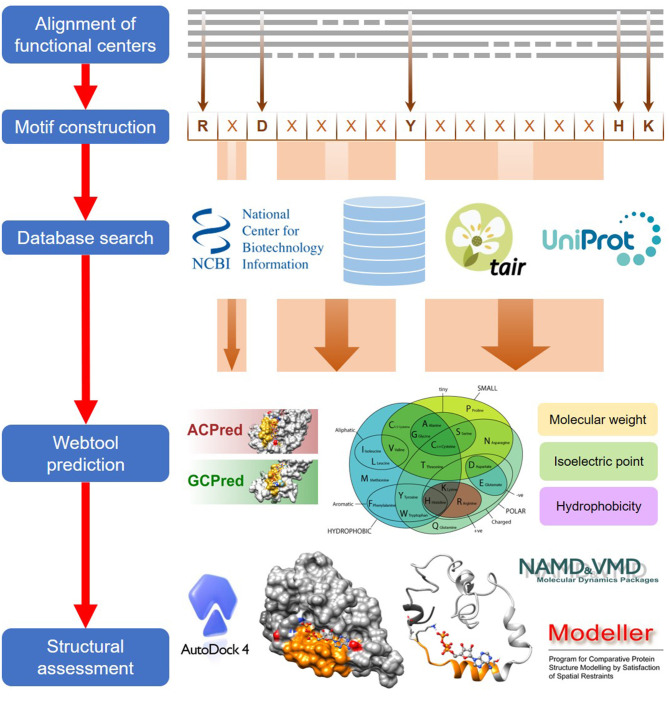
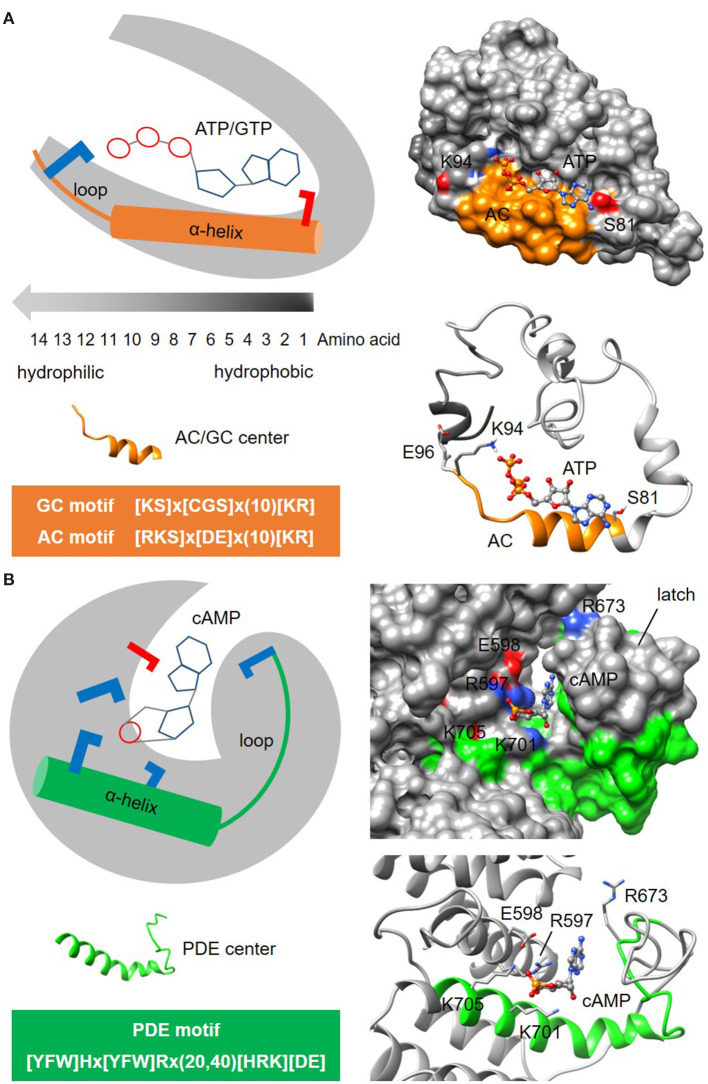
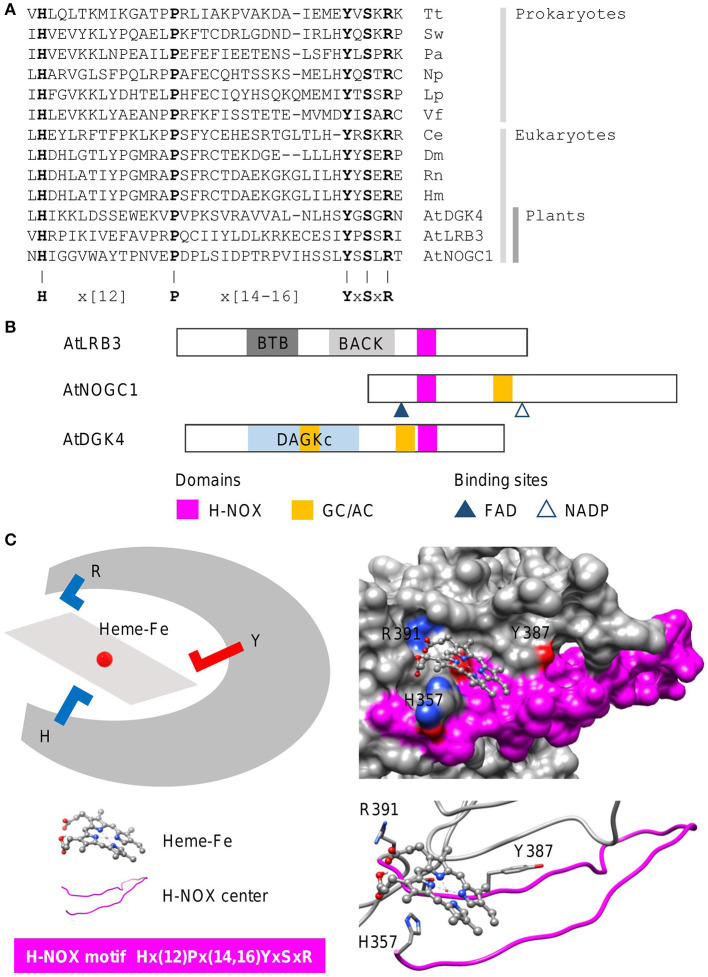
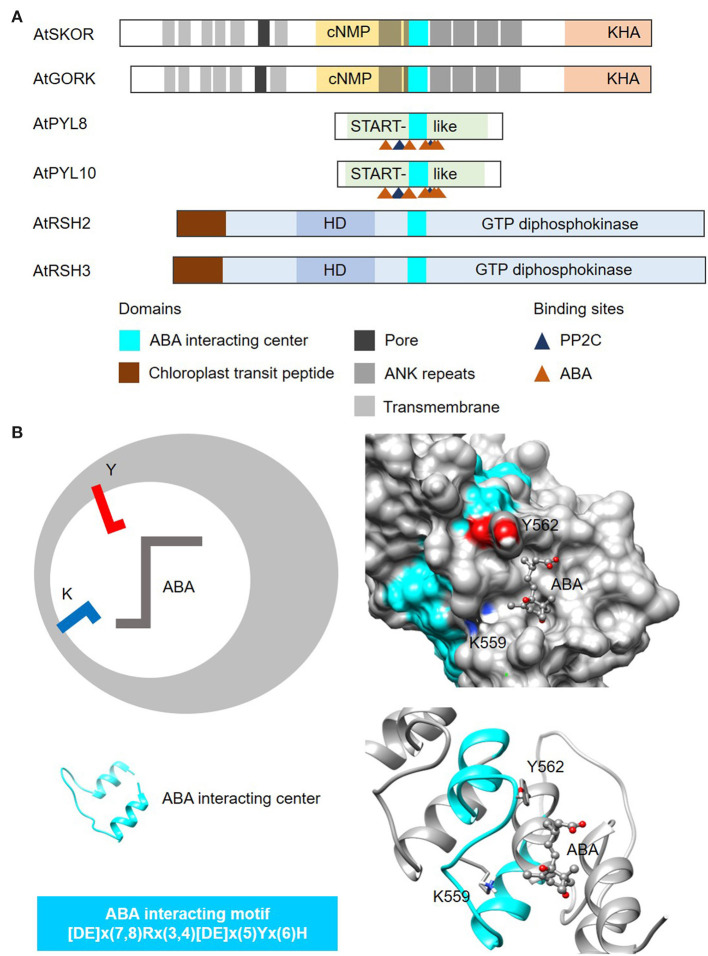
In proteins, functional centers consist of the key amino acids required to perform molecular functions such as catalysis, ligand-binding, hormone- and gas-sensing. These centers are often embedded within complex multi-domain proteins and can perform important cellular signaling functions that enable fine-tuning of temporal and spatial regulation of signaling molecules and networks. To discover hidden functional centers, we have developed a protocol that consists of the following sequential steps. The first is the assembly of a search motif based on the key amino acids in the functional center followed by querying proteomes of interest with the assembled motif. The second consists of a structural assessment of proteins that harbor the motif. This approach, that relies on the application of computational tools for the analysis of data in public repositories and the biological interpretation of the search results, has to-date uncovered several novel functional centers in complex proteins. Here, we use recent examples to describe a step-by-step guide that details the workflow of this approach and supplement with notes, recommendations and cautions to make this protocol robust and widely applicable for the discovery of hidden functional centers.
Keywords: H-NOX; abscisic acid receptor; adenylyl cyclase; functional centers; guanylate cyclase; hidden domains; moonlighting proteins; nitric oxide sensors.
Copyright © 2021 Zhou, Chi, Shen, Dou, Wang, Tian, Gehring and Wong.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Amino acid motifs for the identification of novel protein interactants.Comput Struct Biotechnol J. 2022 Dec 10;21:326-334. doi: 10.1016/j.csbj.2022.12.012. eCollection 2023. Comput Struct Biotechnol J. 2022. PMID: 36582434 Free PMC article. Review.
-
Moonlighting Crypto-Enzymes and Domains as Ancient and Versatile Signaling Devices.Int J Mol Sci. 2024 Sep 2;25(17):9535. doi: 10.3390/ijms25179535. Int J Mol Sci. 2024. PMID: 39273482 Free PMC article. Review.
-
Discovery of Novel Functional Centers With Rationally Designed Amino Acid Motifs.Comput Struct Biotechnol J. 2018 Feb 27;16:70-76. doi: 10.1016/j.csbj.2018.02.007. eCollection 2018. Comput Struct Biotechnol J. 2018. PMID: 29977479 Free PMC article. Review.
-
Conserved Functional Motifs and Homology Modeling to Predict Hidden Moonlighting Functional Sites.Front Bioeng Biotechnol. 2015 Jun 9;3:82. doi: 10.3389/fbioe.2015.00082. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 26106597 Free PMC article. Review.
-
A tandem motif-based and structural approach can identify hidden functional phosphodiesterases.Comput Struct Biotechnol J. 2021 Jan 26;19:970-975. doi: 10.1016/j.csbj.2021.01.036. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 33613864 Free PMC article.
Cited by
-
Amino acid motifs for the identification of novel protein interactants.Comput Struct Biotechnol J. 2022 Dec 10;21:326-334. doi: 10.1016/j.csbj.2022.12.012. eCollection 2023. Comput Struct Biotechnol J. 2022. PMID: 36582434 Free PMC article. Review.
-
Moonlighting Crypto-Enzymes and Domains as Ancient and Versatile Signaling Devices.Int J Mol Sci. 2024 Sep 2;25(17):9535. doi: 10.3390/ijms25179535. Int J Mol Sci. 2024. PMID: 39273482 Free PMC article. Review.
-
New Horizons in Plant Cell Signaling.Int J Mol Sci. 2022 May 23;23(10):5826. doi: 10.3390/ijms23105826. Int J Mol Sci. 2022. PMID: 35628641 Free PMC article.
-
Functional Crypto-Adenylate Cyclases Operate in Complex Plant Proteins.Front Plant Sci. 2021 Aug 12;12:711749. doi: 10.3389/fpls.2021.711749. eCollection 2021. Front Plant Sci. 2021. PMID: 34456950 Free PMC article.
-
"Cyclic nucleotides in plants: from obscure messengers to central regulators".Front Plant Sci. 2025 Jun 25;16:1618243. doi: 10.3389/fpls.2025.1618243. eCollection 2025. Front Plant Sci. 2025. PMID: 40636015 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources