

Cryo-EM Analyses Permit Visualization of Structural Polymorphism of Biological Macromolecules
- PMID: 36303748
- PMCID: PMC9580929
- DOI: 10.3389/fbinf.2021.788308
Cryo-EM Analyses Permit Visualization of Structural Polymorphism of Biological Macromolecules
Abstract
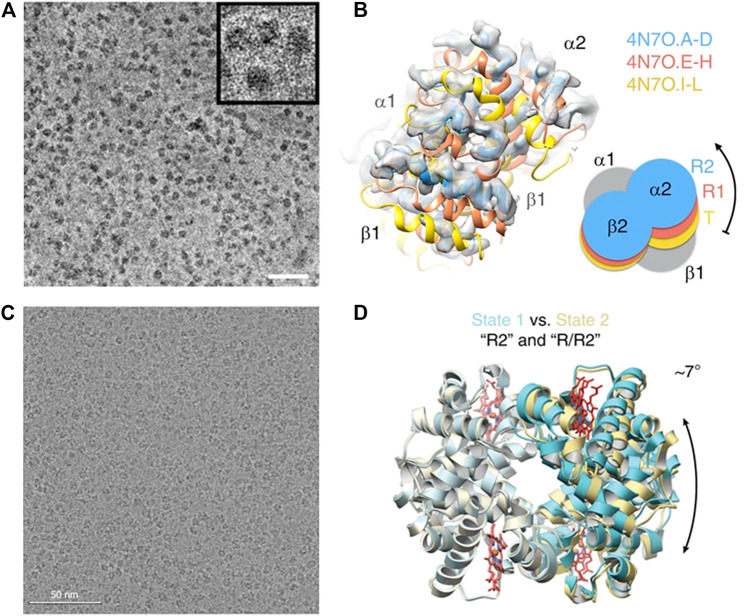
The functions of biological macromolecules are often associated with conformational malleability of the structures. This phenomenon of chemically identical molecules with different structures is coined structural polymorphism. Conventionally, structural polymorphism is observed directly by structural determination at the density map level from X-ray crystal diffraction. Although crystallography approach can report the conformation of a macromolecule with the position of each atom accurately defined in it, the exploration of structural polymorphism and interpreting biological function in terms of crystal structures is largely constrained by the crystal packing. An alternative approach to studying the macromolecule of interest in solution is thus desirable. With the advancement of instrumentation and computational methods for image analysis and reconstruction, cryo-electron microscope (cryo-EM) has been transformed to be able to produce "in solution" structures of macromolecules routinely with resolutions comparable to crystallography but without the need of crystals. Since the sample preparation of single-particle cryo-EM allows for all forms co-existing in solution to be simultaneously frozen, the image data contain rich information as to structural polymorphism. The ensemble of structure information can be subsequently disentangled through three-dimensional (3D) classification analyses. In this review, we highlight important examples of protein structural polymorphism in relation to allostery, subunit cooperativity and function plasticity recently revealed by cryo-EM analyses, and review recent developments in 3D classification algorithms including neural network/deep learning approaches that would enable cryo-EM analyese in this regard. Finally, we brief the frontier of cryo-EM structure determination of RNA molecules where resolving the structural polymorphism is at dawn.
Keywords: classification; conformation alanalysis; cryo-TEM; crystal; heterogeneity; solution; strucural analysis; x-ray.
Copyright © 2021 Chang, Huang, Lin, Chung and Tu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
Publication types
LinkOut - more resources
Full Text Sources