

Viral protein engagement of GBF1 induces host cell vulnerability through synthetic lethality
- PMID: 36305789
- PMCID: PMC9623979
- DOI: 10.1083/jcb.202011050
Viral protein engagement of GBF1 induces host cell vulnerability through synthetic lethality
Abstract
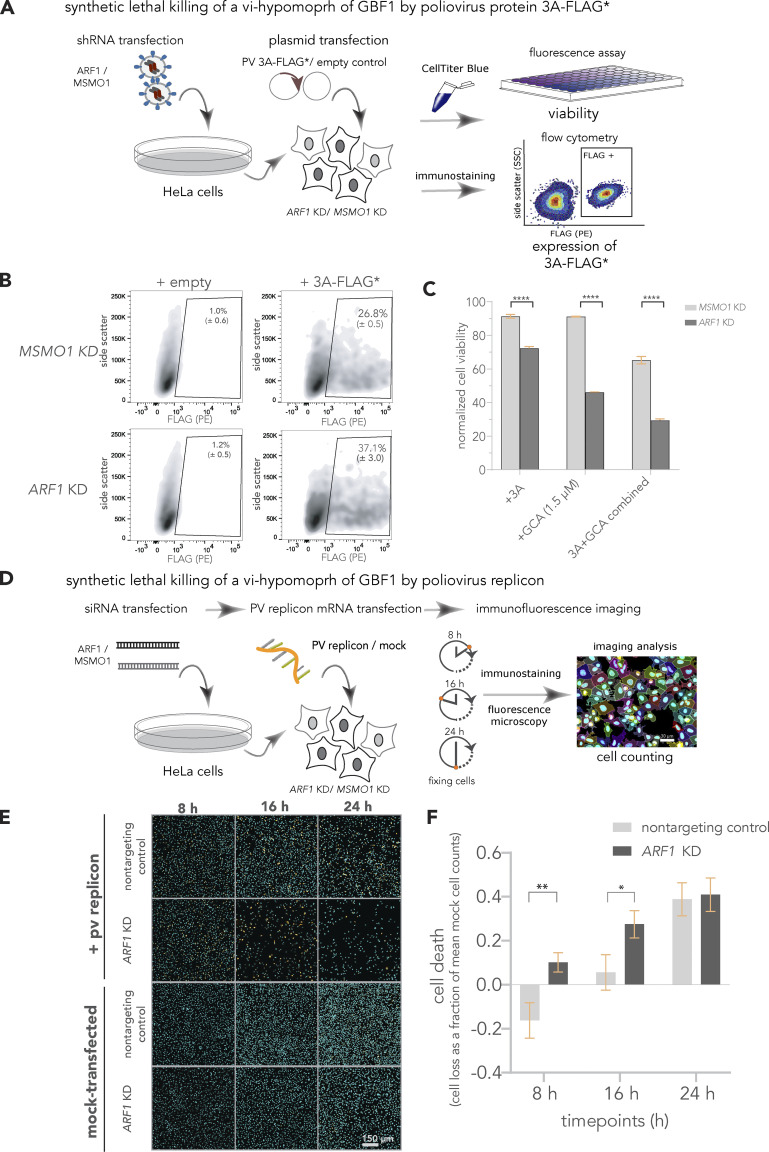
Viruses co-opt host proteins to carry out their lifecycle. Repurposed host proteins may thus become functionally compromised; a situation analogous to a loss-of-function mutation. We term such host proteins as viral-induced hypomorphs. Cells bearing cancer driver loss-of-function mutations have successfully been targeted with drugs perturbing proteins encoded by the synthetic lethal (SL) partners of cancer-specific mutations. Similarly, SL interactions of viral-induced hypomorphs can potentially be targeted as host-based antiviral therapeutics. Here, we use GBF1, which supports the infection of many RNA viruses, as a proof-of-concept. GBF1 becomes a hypomorph upon interaction with the poliovirus protein 3A. Screening for SL partners of GBF1 revealed ARF1 as the top hit, disruption of which selectively killed cells that synthesize 3A alone or in the context of a poliovirus replicon. Thus, viral protein interactions can induce hypomorphs that render host cells selectively vulnerable to perturbations that leave uninfected cells otherwise unscathed. Exploiting viral-induced vulnerabilities could lead to broad-spectrum antivirals for many viruses, including SARS-CoV-2.
© 2022 Navare et al.
Figures






Update of
-
Viral protein engagement of GBF1 induces host cell vulnerability through synthetic lethality.bioRxiv [Preprint]. 2020 Nov 6;221(11):2020.10.12.336487. doi: 10.1101/2020.10.12.336487. bioRxiv. 2020. Update in: J Cell Biol. 2022 Nov 7;221(11):e202011050. doi: 10.1083/jcb.202011050. PMID: 33173868 Free PMC article. Updated. Preprint.
References
-
- Beijersbergen, R.L., Wessels L.F.A., and Bernards R.. 2017. Synthetic lethality in cancer therapeutics. Annu. Rev. Cancer Biol. 1:141–161. 10.1146/annurev-cancerbio-042016-073434 - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
