

Genome-wide detection of human variants that disrupt intronic branchpoints
- PMID: 36306325
- PMCID: PMC9636908
- DOI: 10.1073/pnas.2211194119
Genome-wide detection of human variants that disrupt intronic branchpoints
Abstract
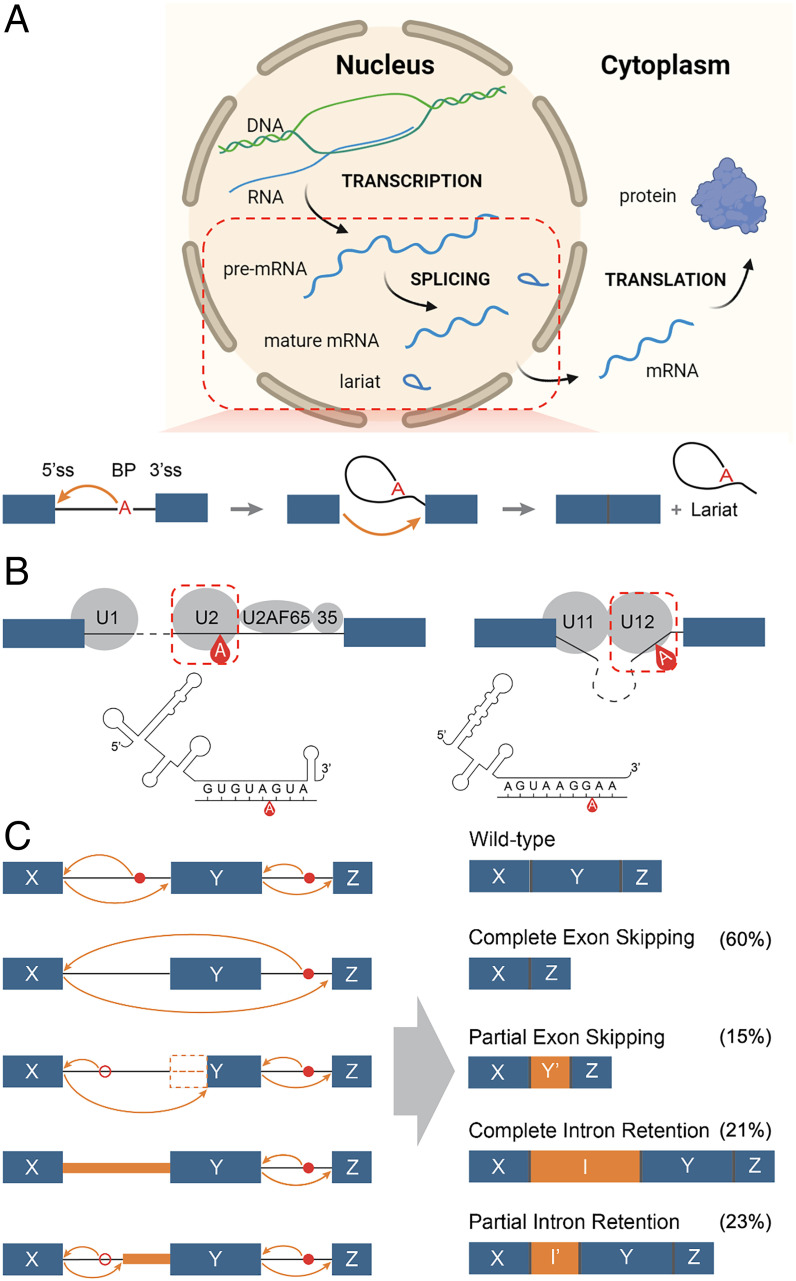
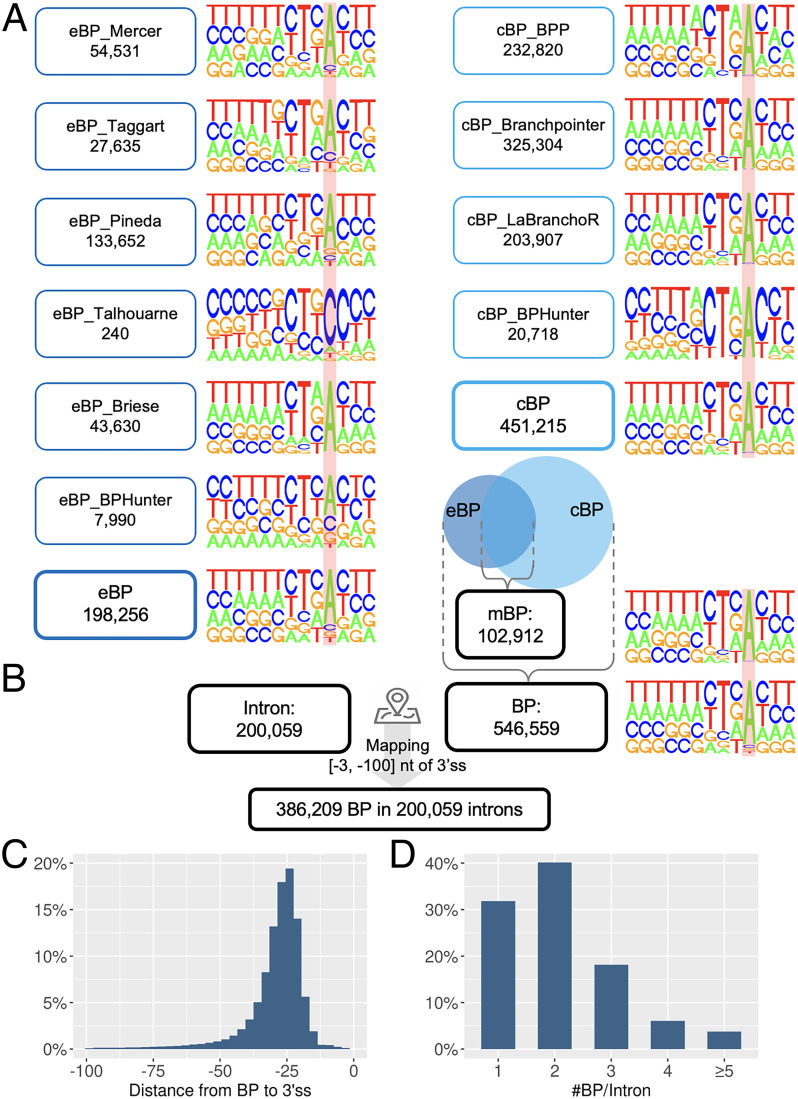
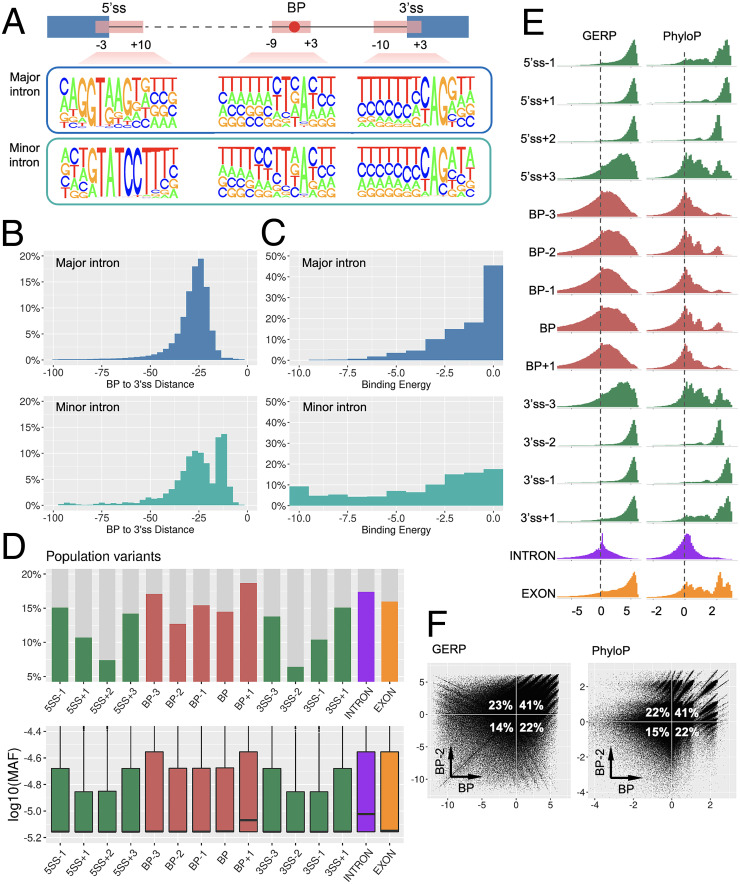
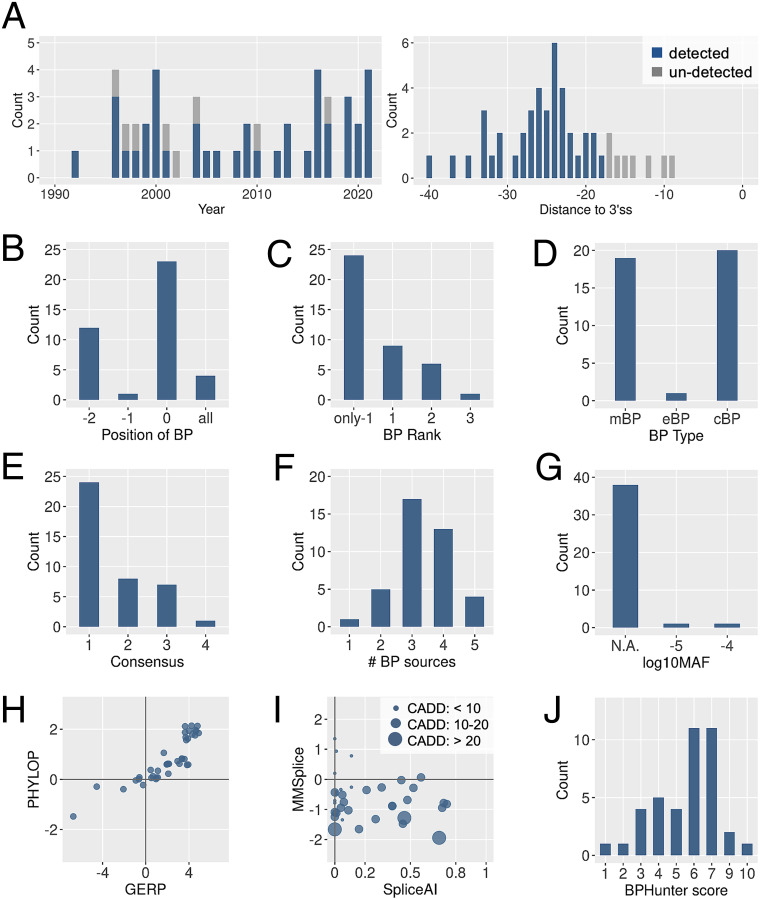
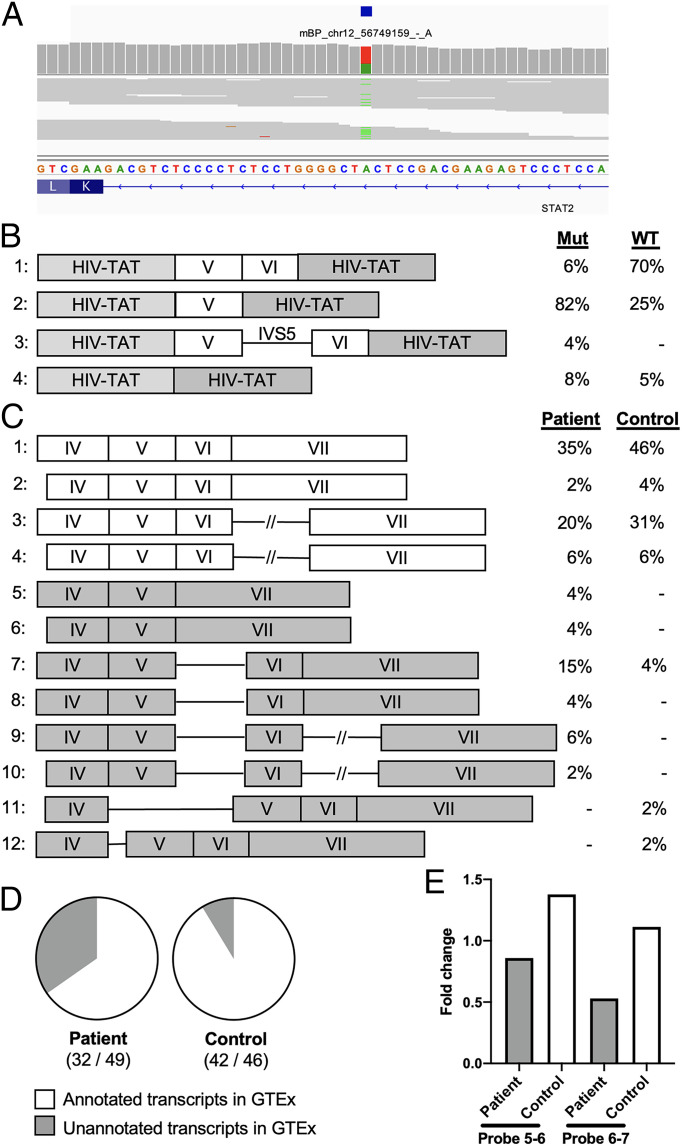
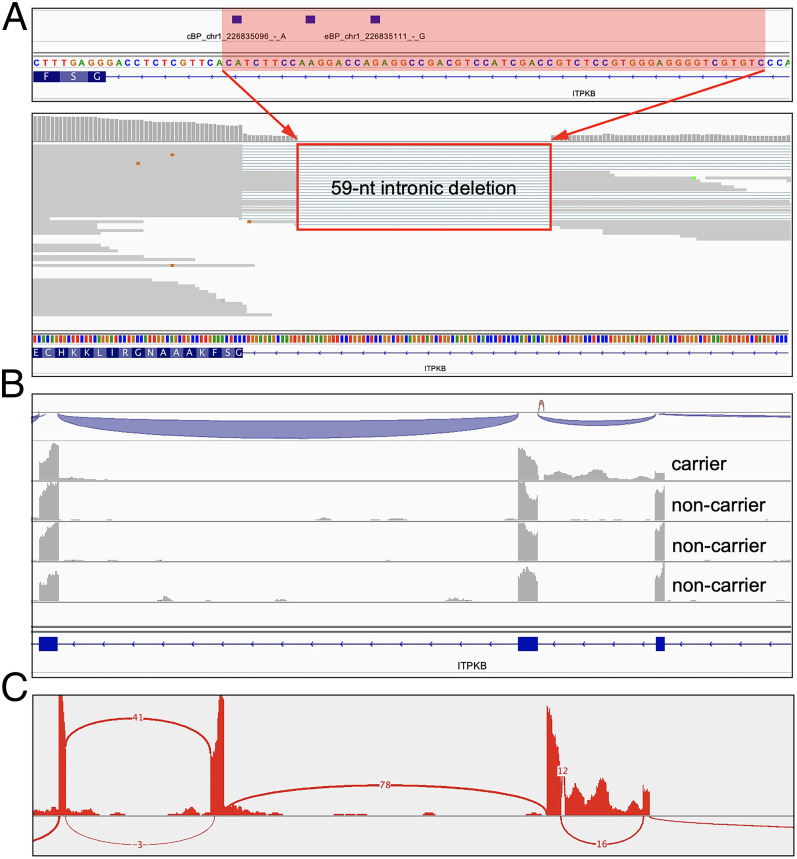
Pre-messenger RNA splicing is initiated with the recognition of a single-nucleotide intronic branchpoint (BP) within a BP motif by spliceosome elements. Forty-eight rare variants in 43 human genes have been reported to alter splicing and cause disease by disrupting BP. However, until now, no computational approach was available to efficiently detect such variants in massively parallel sequencing data. We established a comprehensive human genome-wide BP database by integrating existing BP data and generating new BP data from RNA sequencing of lariat debranching enzyme DBR1-mutated patients and from machine-learning predictions. We characterized multiple features of BP in major and minor introns and found that BP and BP-2 (two nucleotides upstream of BP) positions exhibit a lower rate of variation in human populations and higher evolutionary conservation than the intronic background, while being comparable to the exonic background. We developed BPHunter as a genome-wide computational approach to systematically and efficiently detect intronic variants that may disrupt BP recognition. BPHunter retrospectively identified 40 of the 48 known pathogenic BP variants, in which we summarized a strategy for prioritizing BP variant candidates. The remaining eight variants all create AG-dinucleotides between the BP and acceptor site, which is the likely reason for missplicing. We demonstrated the practical utility of BPHunter prospectively by using it to identify a novel germline heterozygous BP variant of STAT2 in a patient with critical COVID-19 pneumonia and a novel somatic intronic 59-nucleotide deletion of ITPKB in a lymphoma patient, both of which were validated experimentally. BPHunter is publicly available from https://hgidsoft.rockefeller.edu/BPHunter and https://github.com/casanova-lab/BPHunter.
Keywords: branchpoint; disease genetics; intronic variant; software; splicing.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
