

A missense, loss-of-function YARS1 variant in a patient with proximal-predominant motor neuropathy
- PMID: 36307205
- PMCID: PMC9808560
- DOI: 10.1101/mcs.a006246
A missense, loss-of-function YARS1 variant in a patient with proximal-predominant motor neuropathy
Abstract
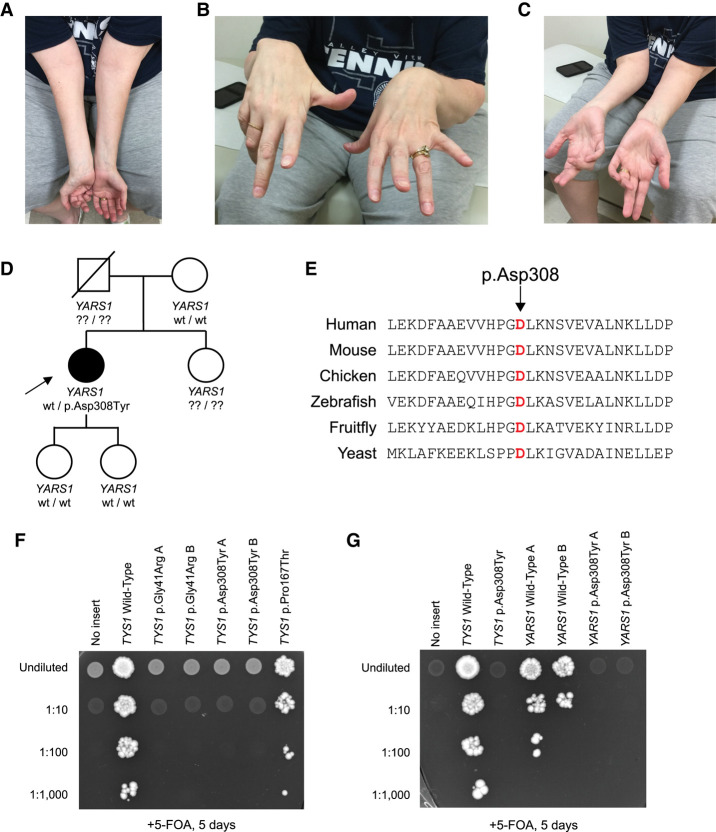
Aminoacyl-tRNA synthetases (ARSs) are essential enzymes with a critical role in protein synthesis: charging tRNA molecules with cognate amino acids. Heterozygosity for variants in five genes (AARS1, GARS1, HARS1, WARS1, and YARS1) encoding cytoplasmic, dimeric ARSs have been associated with autosomal dominant neurological phenotypes, including axonal Charcot-Marie-Tooth disease (CMT). Missense variants in the catalytic domain of YARS1 were previously linked to dominant intermediate CMT type C (DI-CMTC). Here, we report a patient with a missense variant of unknown significance predicted to modify residue 308 in the anticodon binding domain of YARS1 (p.Asp308Tyr). Interestingly, p.Asp308Tyr is associated with proximal-predominant motor neuropathy, which has not been reported in patients with pathogenic YARS1 variants. We demonstrate that this allele causes a loss-of-function effect in yeast complementation assays when modeled in YARS1 and the yeast ortholog TYS1; structural modeling of this variant further supports a loss-of-function effect. Taken together, this study raises the possibility that certain YARS1 variants cause proximal-prominent motor neuropathy and indicates that patients with this phenotype should be screened for genetic lesions in YARS1.
Keywords: areflexia of upper limbs; generalized limb muscle atrophy; lower limb muscle weakness; upper limb muscle weakness.
© 2022 Forrest et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, et al. 2003. Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72: 1293–1299. 10.1086/375039 - DOI - PMC - PubMed
-
- Antonellis A, Lee-Lin SQ, Wasterlain A, Leo P, Quezado M, Goldfarb LG, Myung K, Burgess S, Fischbeck KH, Green ED. 2006. Functional analyses of glycyl-tRNA synthetase mutations suggest a key role for tRNA-charging enzymes in peripheral axons. J Neurosci 26: 10397–10406. 10.1523/JNEUROSCI.1671-06.2006 - DOI - PMC - PubMed
-
- Averdunk L, Sticht H, Surowy H, Lüdecke HJ, Koch-Hogrebe M, Alsaif HS, Kahrizi K, Alzaidan H, Alawam BS, Tohary M, et al. 2021. The recurrent missense mutation p.(Arg367Trp) in YARS1 causes a distinct neurodevelopmental phenotype. J Mol Med (Berl) 99: 1755–1768. 10.1007/s00109-021-02124-9 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases