

The clinical and molecular landscape of congenital myasthenic syndromes in Austria: a nationwide study
- PMID: 36308527
- PMCID: PMC9886627
- DOI: 10.1007/s00415-022-11440-0
The clinical and molecular landscape of congenital myasthenic syndromes in Austria: a nationwide study
Abstract
Background: Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by genetic defects resulting in impaired neuromuscular transmission. Although effective treatments are available, CMS is probably underdiagnosed, and systematic clinico-genetic investigations are warranted.
Methods: We used a nationwide approach to collect Austrian patients with genetically confirmed CMS. We provide a clinical and molecular characterization of this cohort and aimed to ascertain the current frequency of CMS in Austria.
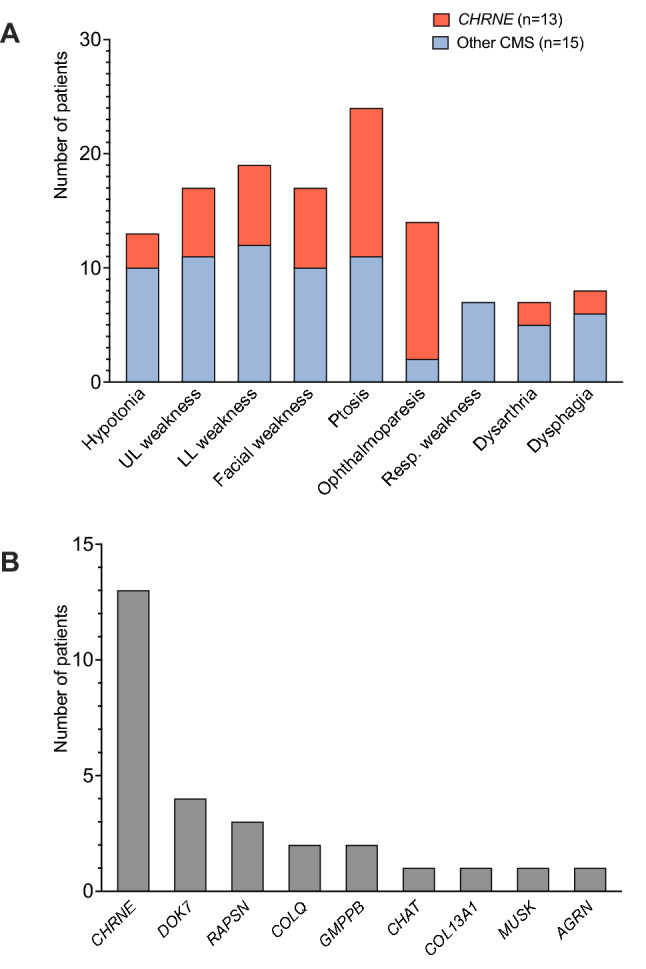
Results: Twenty-eight cases with genetically confirmed CMS were identified, corresponding to an overall prevalence of 3.1 per million (95% CI 2.0-4.3) in Austria. The most frequent genetic etiology was CHRNE (n = 13), accounting for 46.4% of the cohort. Within this subgroup, the variant c.1327del, p.(Glu443Lysfs*64) was detected in nine individuals. Moreover, causative variants were found in DOK7 (n = 4), RAPSN (n = 3), COLQ (n = 2), GMPPB (n = 2), CHAT (n = 1), COL13A1 (n = 1), MUSK (n = 1) and AGRN (n = 1). Clinical onset within the first year of life was reported in one half of the patients. Across all subtypes, the most common symptoms were ptosis (85.7%), lower limb (67.9%), upper limb (60.7%) and facial weakness (60.7%). The majority of patients (96.4%) received specific treatment, including acetylcholinesterase inhibitors in 20, adrenergic agonists in 11 and 3,4-diaminopyridine in nine patients.
Conclusions: Our study presents the first systematic characterization of individuals with CMS in Austria, providing prevalence estimates and genotype-phenotype correlations that may help to improve the diagnostic approach and patient management.
Keywords: Austria; CHRNE; Congenital myasthenic syndrome; Myasthenia.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
