

Integrator endonuclease drives promoter-proximal termination at all RNA polymerase II-transcribed loci
- PMID: 36309014
- PMCID: PMC9680917
- DOI: 10.1016/j.molcel.2022.10.004
Integrator endonuclease drives promoter-proximal termination at all RNA polymerase II-transcribed loci
Abstract
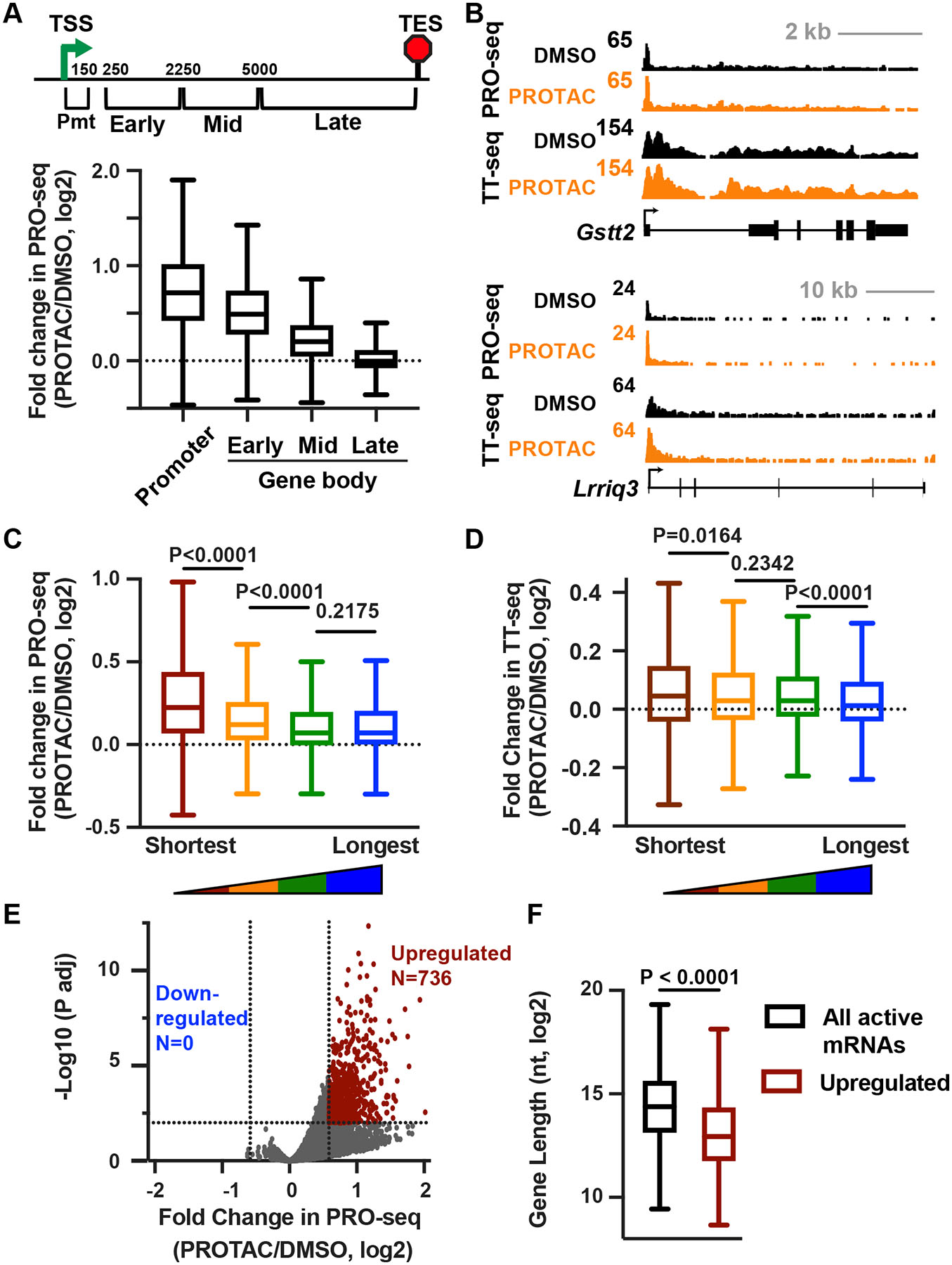
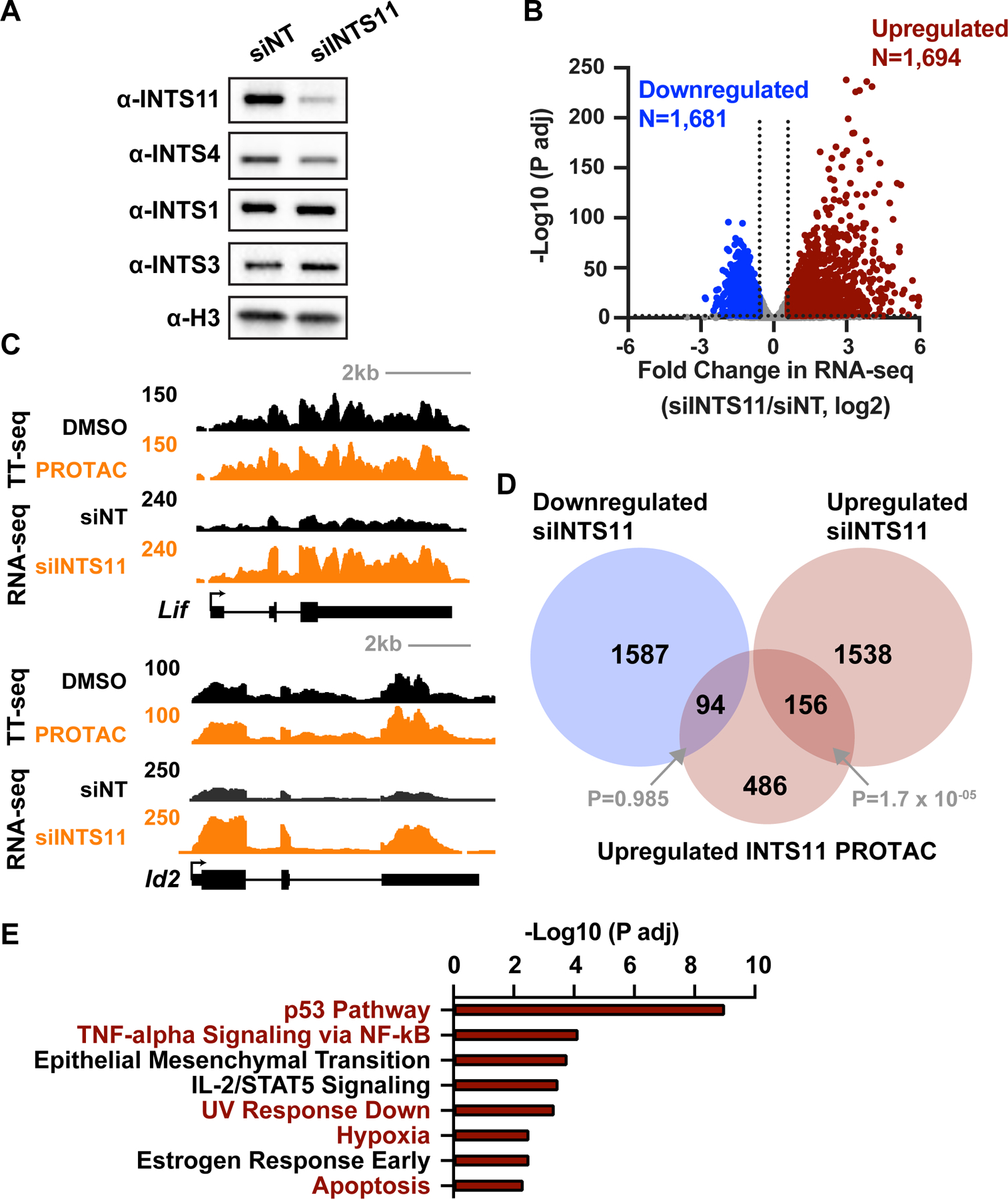
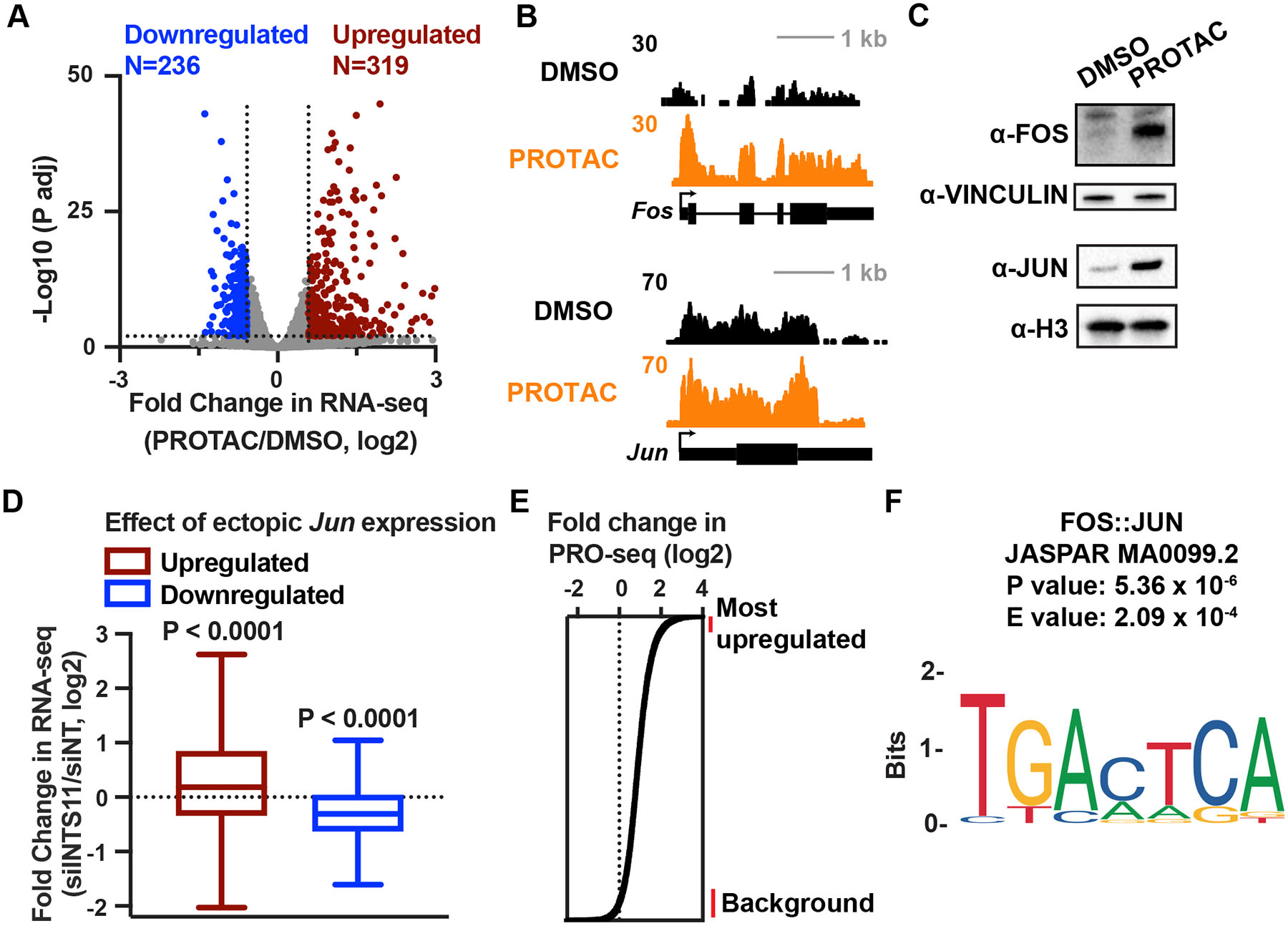
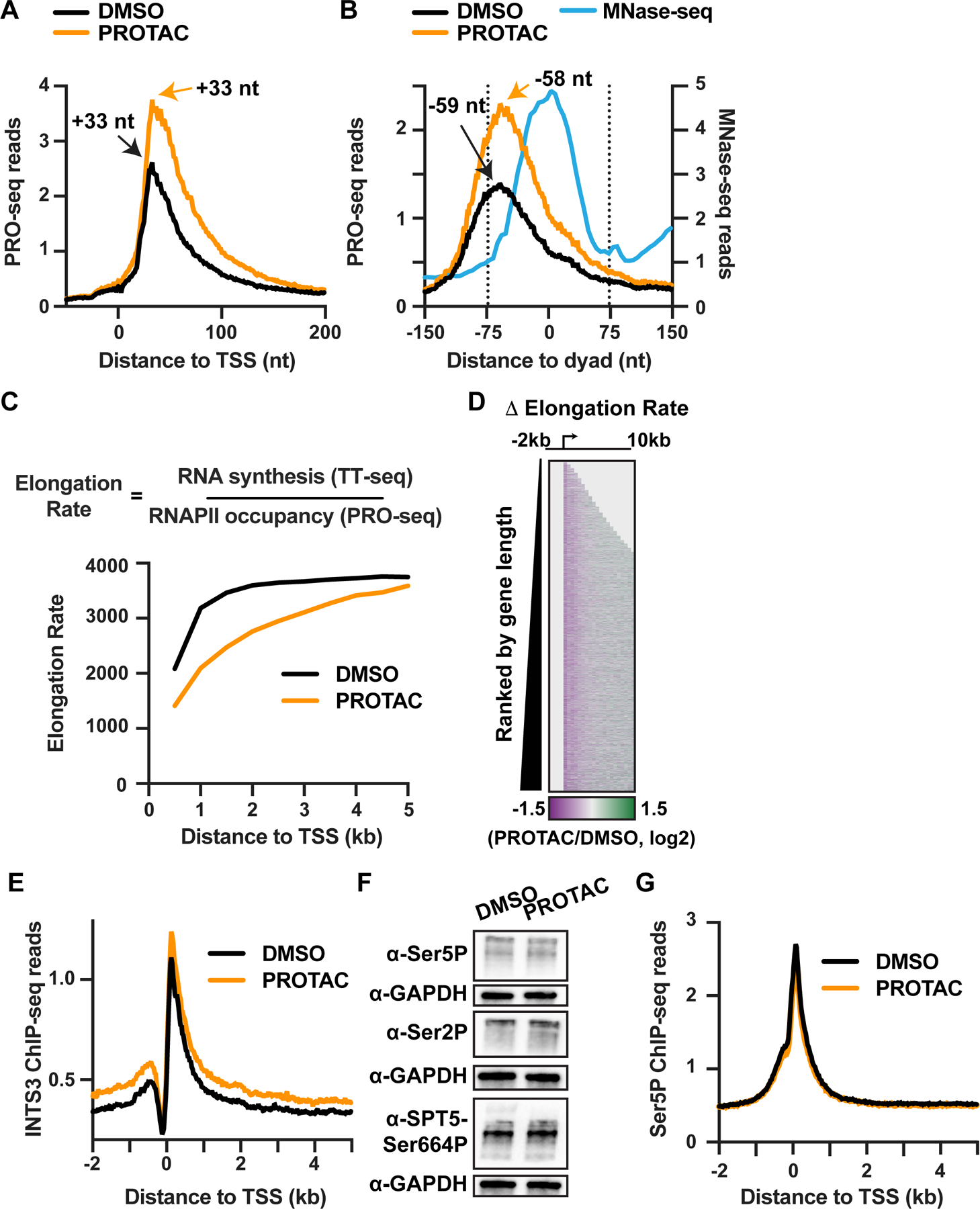
RNA polymerase II (RNAPII) pausing in early elongation is critical for gene regulation. Paused RNAPII can be released into productive elongation by the kinase P-TEFb or targeted for premature termination by the Integrator complex. Integrator comprises endonuclease and phosphatase activities, driving termination by cleavage of nascent RNA and removal of stimulatory phosphorylation. We generated a degron system for rapid Integrator endonuclease (INTS11) depletion to probe the direct consequences of Integrator-mediated RNA cleavage. Degradation of INTS11 elicits nearly universal increases in active early elongation complexes. However, these RNAPII complexes fail to achieve optimal elongation rates and exhibit persistent Integrator phosphatase activity. Thus, only short transcripts are significantly upregulated following INTS11 loss, including transcription factors, signaling regulators, and non-coding RNAs. We propose a uniform molecular function for INTS11 across all RNAPII-transcribed loci, with differential effects on particular genes, pathways, or RNA biotypes reflective of transcript lengths rather than specificity of Integrator activity.
Keywords: Integrator; gene regulation; non-coding RNAs; premature termination; transcription.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests K.A. received research funding from Novartis not related to this work, is on the SAB of CAMP4 Therapeutics, and is a member of the Advisory Board of Molecular Cell.
Figures



References
-
- Baillat D, Hakimi M-A, Näär AM, Shilatifard A, Cooch N, and Shiekhattar R (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
