

Best genome sequencing strategies for annotation of complex immune gene families in wildlife
- PMID: 36310247
- PMCID: PMC9618407
- DOI: 10.1093/gigascience/giac100
Best genome sequencing strategies for annotation of complex immune gene families in wildlife
Abstract
Background: The biodiversity crisis and increasing impact of wildlife disease on animal and human health provides impetus for studying immune genes in wildlife. Despite the recent boom in genomes for wildlife species, immune genes are poorly annotated in nonmodel species owing to their high level of polymorphism and complex genomic organisation. Our research over the past decade and a half on Tasmanian devils and koalas highlights the importance of genomics and accurate immune annotations to investigate disease in wildlife. Given this, we have increasingly been asked the minimum levels of genome quality required to effectively annotate immune genes in order to study immunogenetic diversity. Here we set out to answer this question by manually annotating immune genes in 5 marsupial genomes and 1 monotreme genome to determine the impact of sequencing data type, assembly quality, and automated annotation on accurate immune annotation.
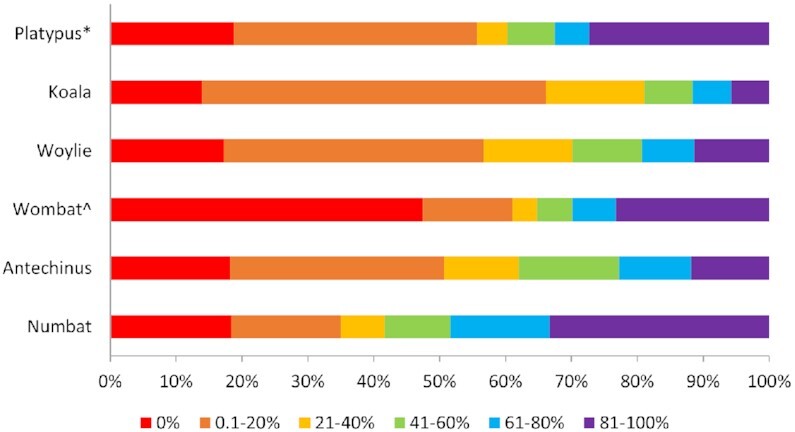
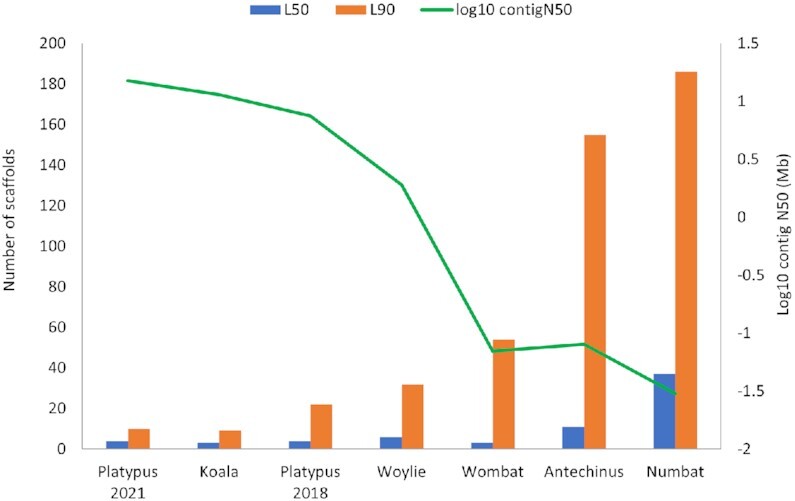
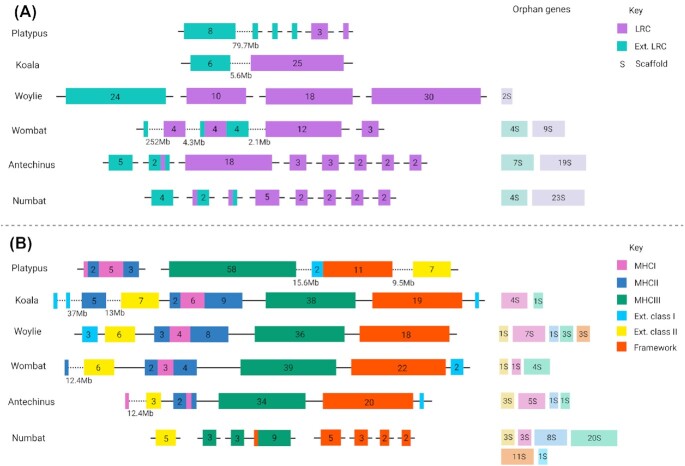
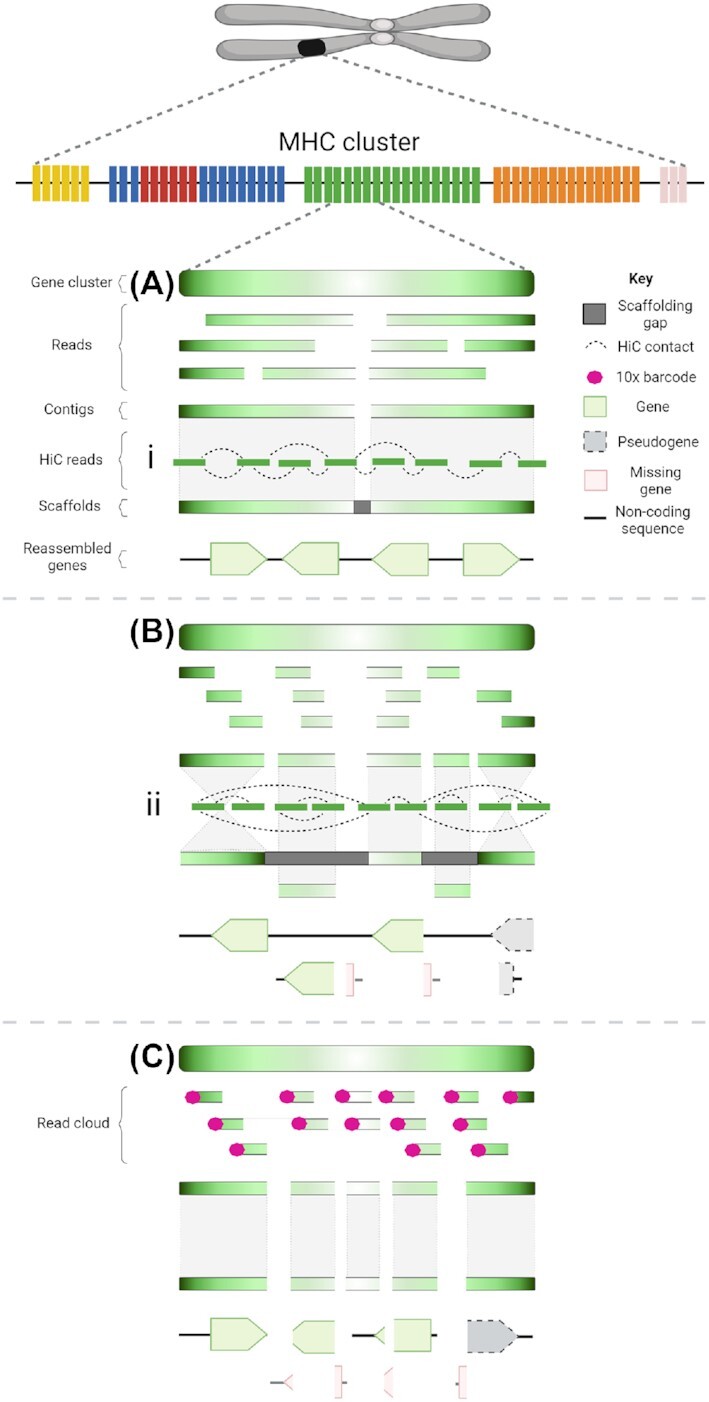
Results: Genome quality is directly linked to our ability to annotate complex immune gene families, with long reads and scaffolding technologies required to reassemble immune gene clusters and elucidate evolution, organisation, and true gene content of the immune repertoire. Draft-quality genomes generated from short reads with HiC or 10× Chromium linked reads were unable to achieve this. Despite mammalian BUSCOv5 scores of up to 94.1% amongst the 6 genomes, automated annotation pipelines incorrectly annotated up to 59% of manually annotated immune genes regardless of assembly quality or method of automated annotation.
Conclusions: Our results demonstrate that long reads and scaffolding technologies, alongside manual annotation, are required to accurately study the immune gene repertoire of wildlife species.
Keywords: MHC; annotation; disease; genome; immune gene; quality; wildlife.
© The Author(s) 2022. Published by Oxford University Press GigaScience.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing.BMC Genomics. 2019 Jan 9;20(1):23. doi: 10.1186/s12864-018-5381-7. BMC Genomics. 2019. PMID: 30626323 Free PMC article.
-
Plethora of New Marsupial Genomes Informs Our Knowledge of Marsupial MHC Class II.Genome Biol Evol. 2024 Aug 5;16(8):evae156. doi: 10.1093/gbe/evae156. Genome Biol Evol. 2024. PMID: 39031605 Free PMC article.
-
Structural and functional annotation of the porcine immunome.BMC Genomics. 2013 May 15;14:332. doi: 10.1186/1471-2164-14-332. BMC Genomics. 2013. PMID: 23676093 Free PMC article.
-
Scalable and versatile container-based pipelines for de novo genome assembly and bacterial annotation.F1000Res. 2023 Sep 25;12:1205. doi: 10.12688/f1000research.139488.1. eCollection 2023. F1000Res. 2023. PMID: 37970066 Free PMC article. Review.
-
A beginner's guide to eukaryotic genome annotation.Nat Rev Genet. 2012 Apr 18;13(5):329-42. doi: 10.1038/nrg3174. Nat Rev Genet. 2012. PMID: 22510764 Review.
Cited by
-
Genome-wide diversity and MHC characterisation in a critically endangered freshwater turtle susceptible to disease.Immunogenetics. 2025 May 6;77(1):21. doi: 10.1007/s00251-025-01378-8. Immunogenetics. 2025. PMID: 40327086 Free PMC article.
-
Marsupial cathelicidins: characterization, antimicrobial activity and evolution in this unique mammalian lineage.Front Immunol. 2025 Apr 4;16:1524092. doi: 10.3389/fimmu.2025.1524092. eCollection 2025. Front Immunol. 2025. PMID: 40255401 Free PMC article.
-
Using bioinformatics to investigate functional diversity: a case study of MHC diversity in koalas.Immunogenetics. 2024 Dec;76(5-6):381-395. doi: 10.1007/s00251-024-01356-6. Epub 2024 Oct 5. Immunogenetics. 2024. PMID: 39367971 Free PMC article.
-
A genome assembly and transcriptome atlas of the inbred Babraham pig to illuminate porcine immunogenetic variation.Immunogenetics. 2024 Dec;76(5-6):361-380. doi: 10.1007/s00251-024-01355-7. Epub 2024 Sep 19. Immunogenetics. 2024. PMID: 39294478 Free PMC article.
-
ToxCodAn-Genome: an automated pipeline for toxin-gene annotation in genome assembly of venomous lineages.Gigascience. 2024 Jan 2;13:giad116. doi: 10.1093/gigascience/giad116. Gigascience. 2024. PMID: 38241143 Free PMC article.
References
-
- Diaz S, Settle J, Brondizio ES, et al. Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. Bonn, Germany: IPBES; 2019.
-
- Scheele BC, Pasmans F, Skerratt LF, et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science. 2019;363(6434):1459. - PubMed
-
- Hoyt JR, Kilpatrick AM, Langwig KE. Ecology and impacts of white-nose syndrome on bats. Nat Rev Microbiol. 2021;19(3):196–210. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
