

Mechanism of Visible Light-Mediated Alkene Aminoarylation with Arylsulfonylacetamides
- PMID: 36312445
- PMCID: PMC9608985
- DOI: 10.1021/acscatal.2c02577
Mechanism of Visible Light-Mediated Alkene Aminoarylation with Arylsulfonylacetamides
Abstract
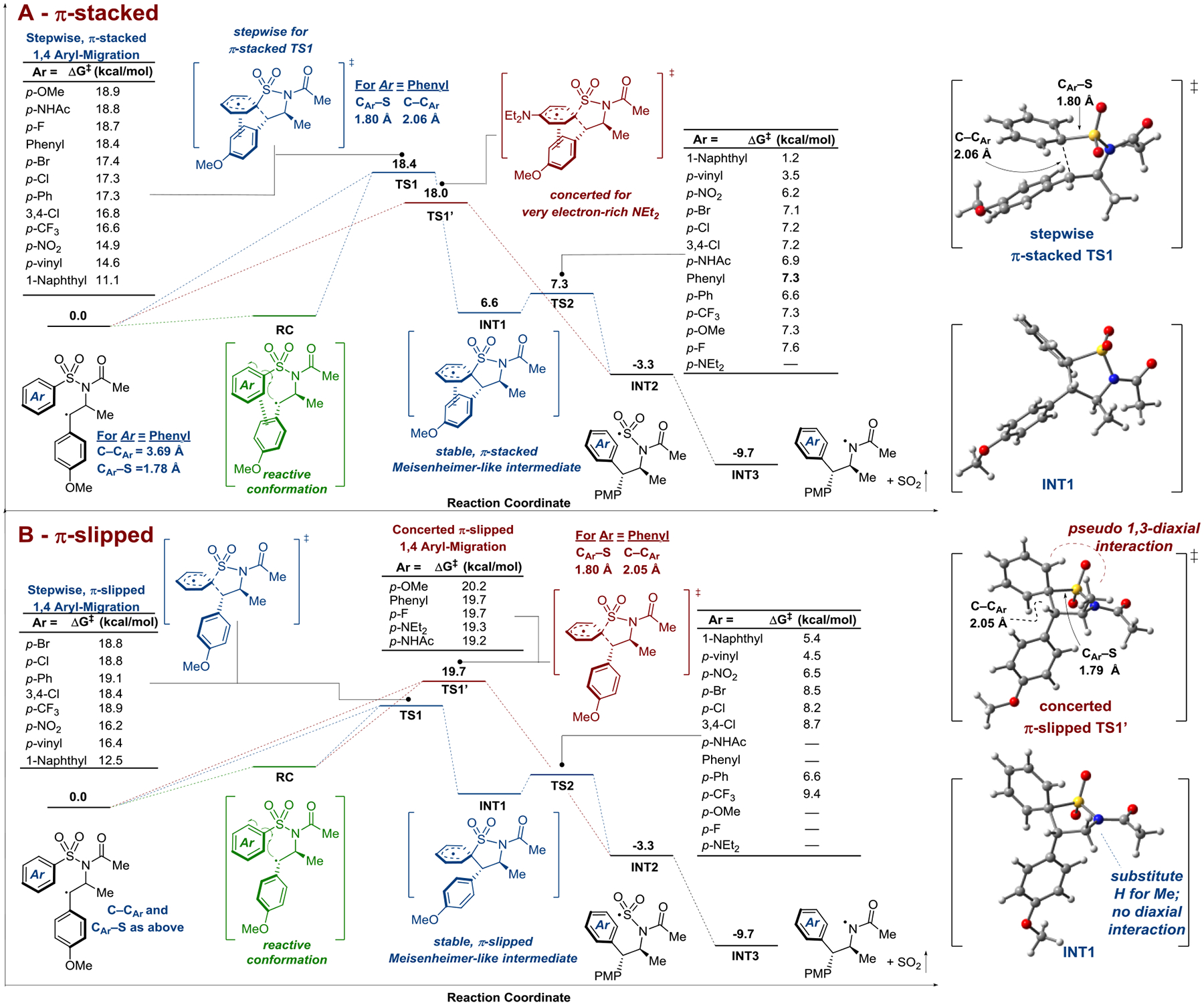
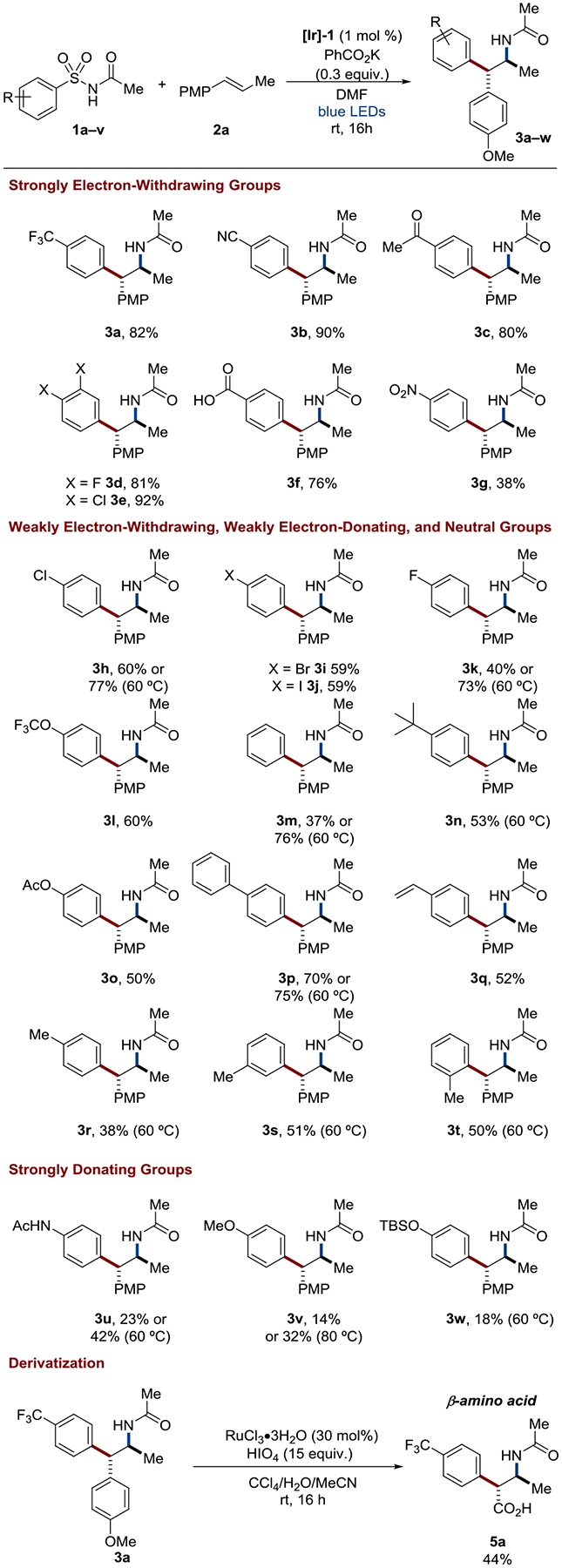
Alkene aminoarylation with arylsulfonylacetamides via a visible-light mediated radical Smiles-Truce rearrangement represents a convenient approach to the privileged arylethylamine pharmacaphore traditionally generated by circuitous, multi-step sequences. Herein, we report detailed synthetic, spectroscopic, kinetic, and computational studies designed to interrogate the proposed mechanism, including the key aryl transfer event. The data are consistent with a rate-limiting 1,4-aryl migration occurring either via a stepwise process involving a radical Meisenheimer-like intermediate or in a concerted fashion dependent on both arene electronics and alkene sterics. Our efforts to probe the mechanism have significantly expanded the substrate scope of the transformation with respect to the migrating aryl group and provide further credence to the synthetic potential of radical aryl migrations.
Keywords: Photoredox; aminoarylation; aryl transfer; nucleophilic aromatic substitution; radical cation.
Conflict of interest statement
The authors declare no competing financial interest.
Figures










References
-
- Nichols DE Amphetamine and Its Analogues; Psychopharmacology, Toxicology, and Abuse. In Amphetamine and Its Analogues; Psychopharmacology, Toxicology, and Abuse; Academic Press: San Diego, CA, 1994; pp 3–41.
-
- Gallardo-Godoy A; Fierro A; McLean TH; Castillo M; Cassels BK; Reyes-Parada M; Nichols DE Sulfur-Substituted α-Alkyl Phenethylamines as Selective and Reversible MAO-A Inhibitors: Biological Activities, CoMFA Analysis, and Active Site Modeling. J. Med. Chem 2005, 48 (7), 2407–2419. - PubMed
-
- Reti L β-Phenethylamines. In The Alkaloids: Chemistry and Physiology; Manske RHF, Holmes HL, Eds.; Academic Press, 1953; Vol. 3, pp 313–338.
-
- Claes L; Janssen M; De Vos DE Organocatalytic Decarboxylation of Amino Acids as a Route to Bio-based Amines and Amides. ChemCatChem 2019, 11 (17), 4297–4306.
Grants and funding
LinkOut - more resources
Full Text Sources