

Highly diverse ribonucleic acid viruses in the viromes of eukaryotic host species in Yunnan province, China
- PMID: 36312977
- PMCID: PMC9606678
- DOI: 10.3389/fmicb.2022.1019444
Highly diverse ribonucleic acid viruses in the viromes of eukaryotic host species in Yunnan province, China
Abstract
Background: The diversity in currently documented viruses and their morphological characteristics indicates the need for understanding the evolutionary characteristics of viruses. Notably, further studies are needed to obtain a comprehensive landscape of virome, the virome of host species in Yunnan province, China.
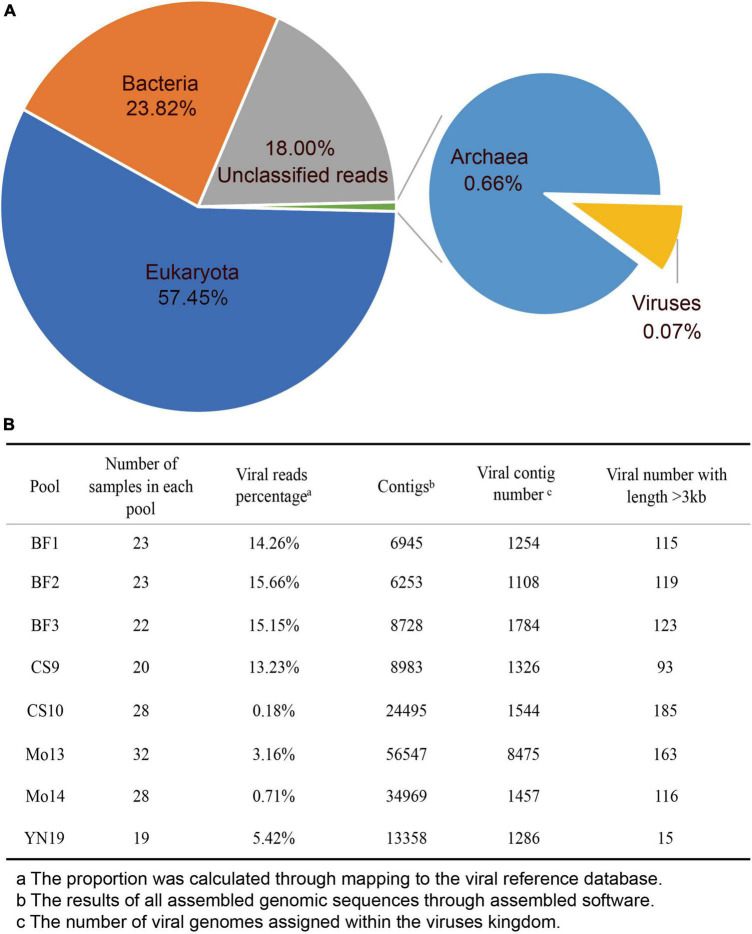
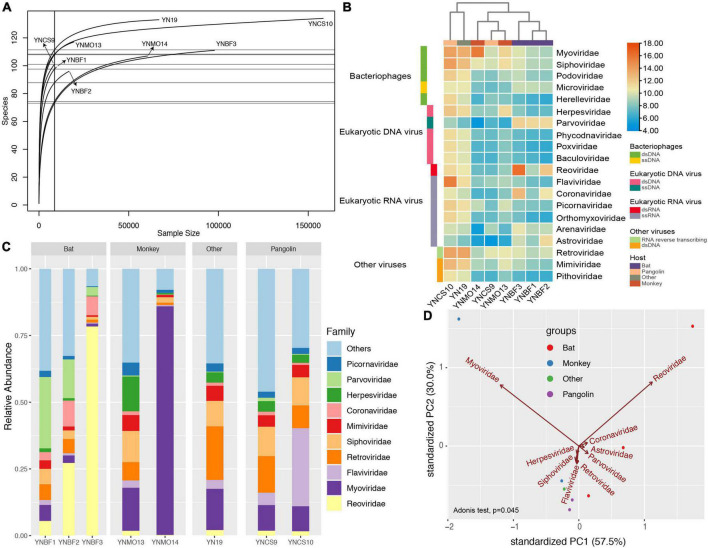
Materials and methods: We implemented the metagenomic next-generation sequencing strategy to investigate the viral diversity, which involved in 465 specimens collected from bats, pangolins, monkeys, and other species. The diverse RNA viruses were analyzed, especially focusing on the genome organization, genetic divergence and phylogenetic relationships.
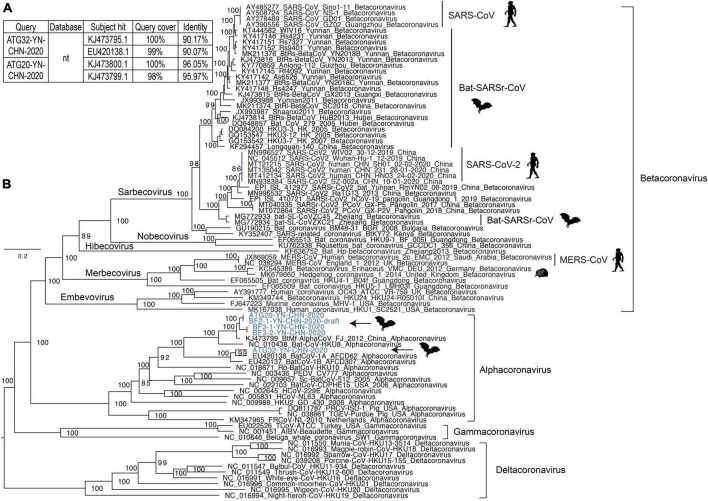
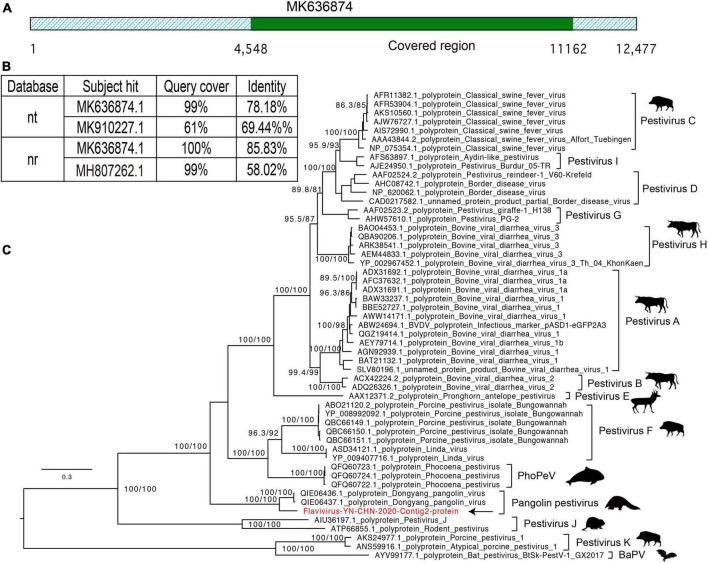
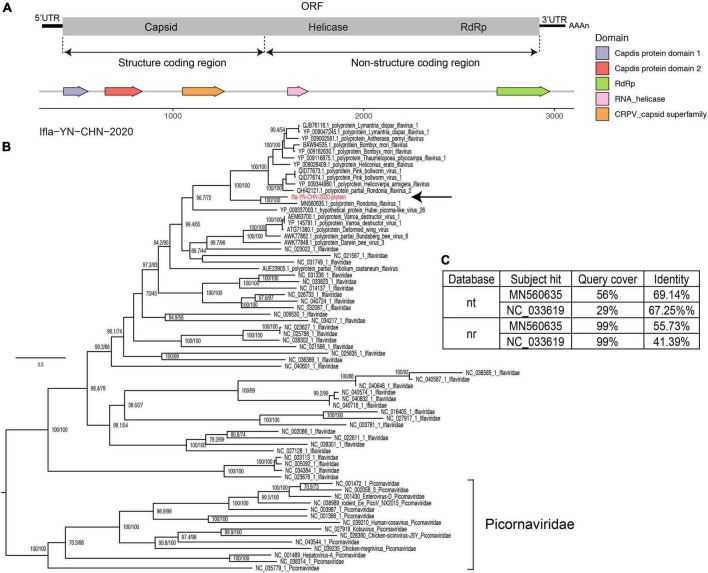
Results: In this study, we investigated the viral composition of eight libraries from bats, pangolins, monkeys, and other species, and found several diverse RNA viruses, including the Alphacoronavirus from bat specimens. By characterizing the genome organization, genetic divergence, and phylogenetic relationships, we identified five Alphacoronavirus strains, which shared phylogenetic association with Bat-CoV-HKU8-related strains. The pestivirus-like virus related to recently identified Dongyang pangolin virus (DYPV) strains from dead pangolin specimens, suggesting that these viruses are evolving. Some genomes showed higher divergence from known species (e.g., calicivirus CS9-Cali-YN-CHN-2020), and many showed evidence of recombination events with unknown or known strains (e.g., mamastroviruses BF2-astro-YN-CHN-2020 and EV-A122 AKM5-YN-CHN-2020). The newly identified viruses showed extensive changes and could be assigned as new species, or even genus (e.g., calicivirus CS9-Cali-YN-CHN-2020 and iflavirus Ifla-YN-CHN-2020). Moreover, we identified several highly divergent RNA viruses and estimated their evolutionary characteristics among different hosts, providing data for further examination of their evolutionary dynamics.
Conclusion: Overall, our study emphasizes the close association between emerging viruses and infectious diseases, and the need for more comprehensive surveys.
Keywords: RNA viruses; coronavirus; metagenomic next-generation sequencing (mNGS); picornavirus; viral evolution; virome.
Copyright © 2022 Han, Xiao, Song, Zhao, Sun, Lu, Zhang, Li, Li, Si, Zhang, Zhao, Jia, Zhou, Wang, Zhu, Yan, Xu, Fu and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Myotis fimbriatus Virome, a Window to Virus Diversity and Evolution in the Genus Myotis.Viruses. 2022 Aug 27;14(9):1899. doi: 10.3390/v14091899. Viruses. 2022. PMID: 36146706 Free PMC article.
-
Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host.Braz J Microbiol. 2020 Dec;51(4):1593-1599. doi: 10.1007/s42770-020-00321-1. Epub 2020 Jun 26. Braz J Microbiol. 2020. PMID: 32592038 Free PMC article.
-
Virome of Bat Guano from Nine Northern California Roosts.J Virol. 2021 Jan 13;95(3):e01713-20. doi: 10.1128/JVI.01713-20. Print 2021 Jan 13. J Virol. 2021. PMID: 33115864 Free PMC article.
-
Evidence for an Ancestral Association of Human Coronavirus 229E with Bats.J Virol. 2015 Dec;89(23):11858-70. doi: 10.1128/JVI.01755-15. Epub 2015 Sep 16. J Virol. 2015. PMID: 26378164 Free PMC article.
-
Diversity and evolution of the animal virome.Nat Rev Microbiol. 2022 Jun;20(6):321-334. doi: 10.1038/s41579-021-00665-x. Epub 2022 Jan 4. Nat Rev Microbiol. 2022. PMID: 34983966 Review.
Cited by
-
Multiple novel caliciviruses identified from stoats (Mustela erminea) in the United Kingdom.Access Microbiol. 2024 Jul 9;6(7):000813.v4. doi: 10.1099/acmi.0.000813.v4. eCollection 2024. Access Microbiol. 2024. PMID: 39130737 Free PMC article.
-
Phylogenetic Analysis and Codon Usage Bias Reveal the Base of Feline and Canine Chaphamaparvovirus for Cross-Species Transmission.Animals (Basel). 2023 Aug 14;13(16):2617. doi: 10.3390/ani13162617. Animals (Basel). 2023. PMID: 37627409 Free PMC article.
-
Identification and characterization of novel bat coronaviruses in Spain.PLoS Pathog. 2025 Aug 4;21(8):e1013371. doi: 10.1371/journal.ppat.1013371. eCollection 2025 Aug. PLoS Pathog. 2025. PMID: 40758759 Free PMC article.
References
LinkOut - more resources
Full Text Sources