

Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex
- PMID: 36314080
- PMCID: PMC10115353
- DOI: 10.1093/brain/awac404
Temporal lobe epilepsy with GAD antibodies: neurons killed by T cells not by complement membrane attack complex
Abstract
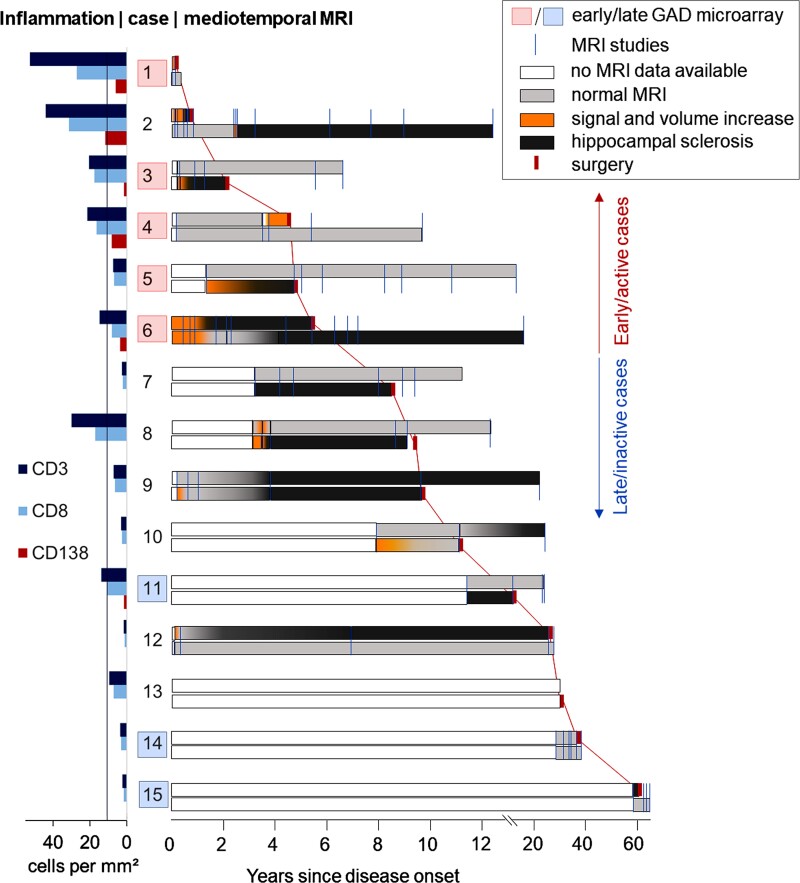
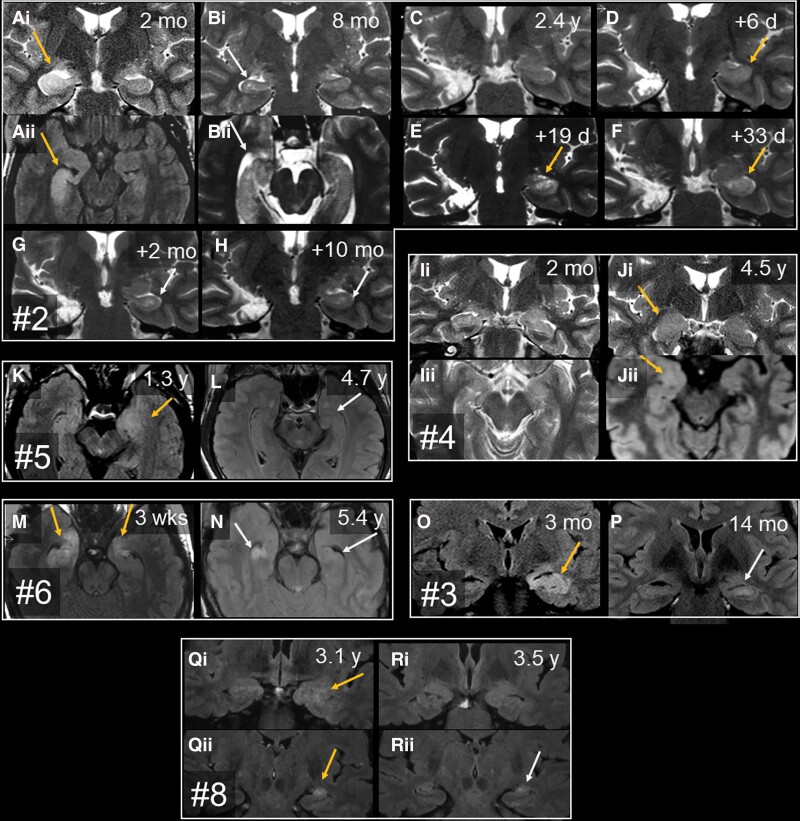
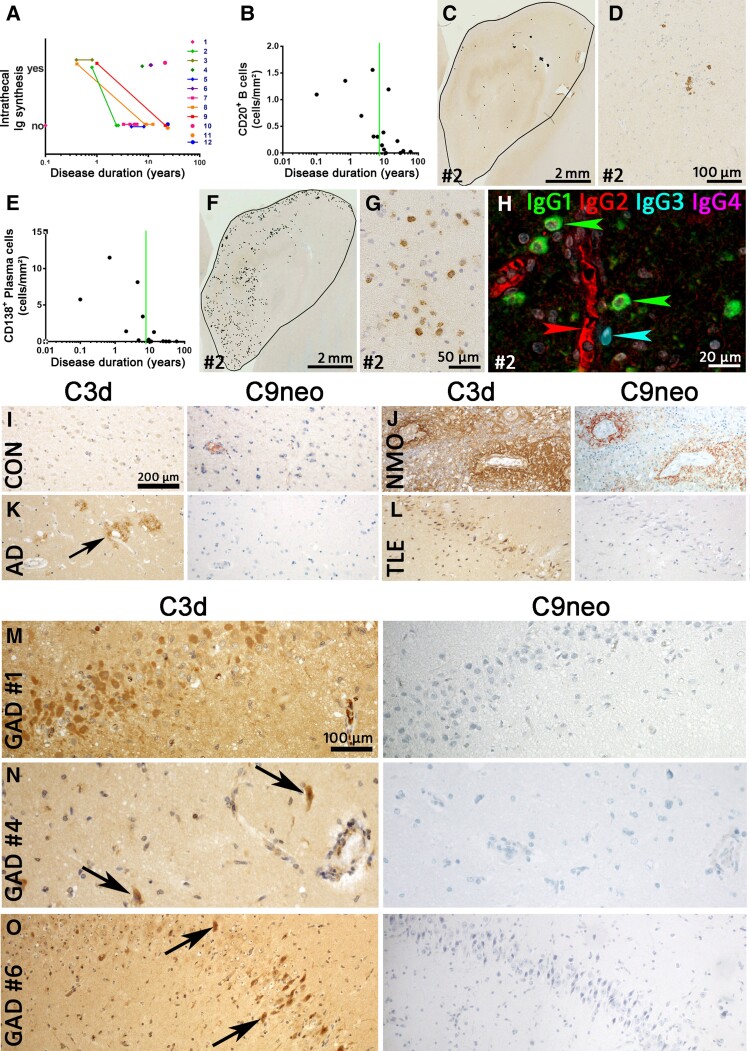
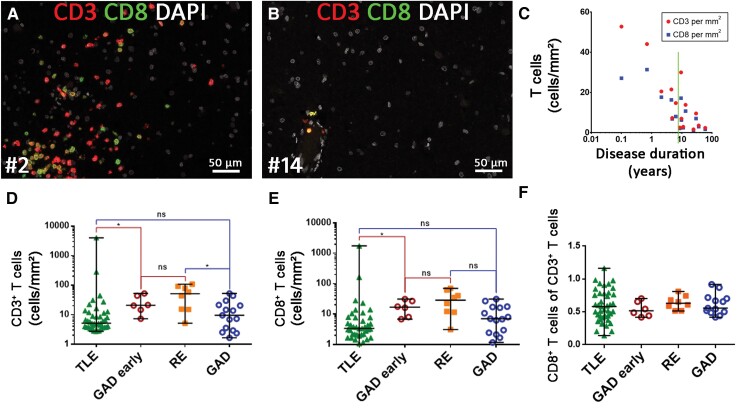
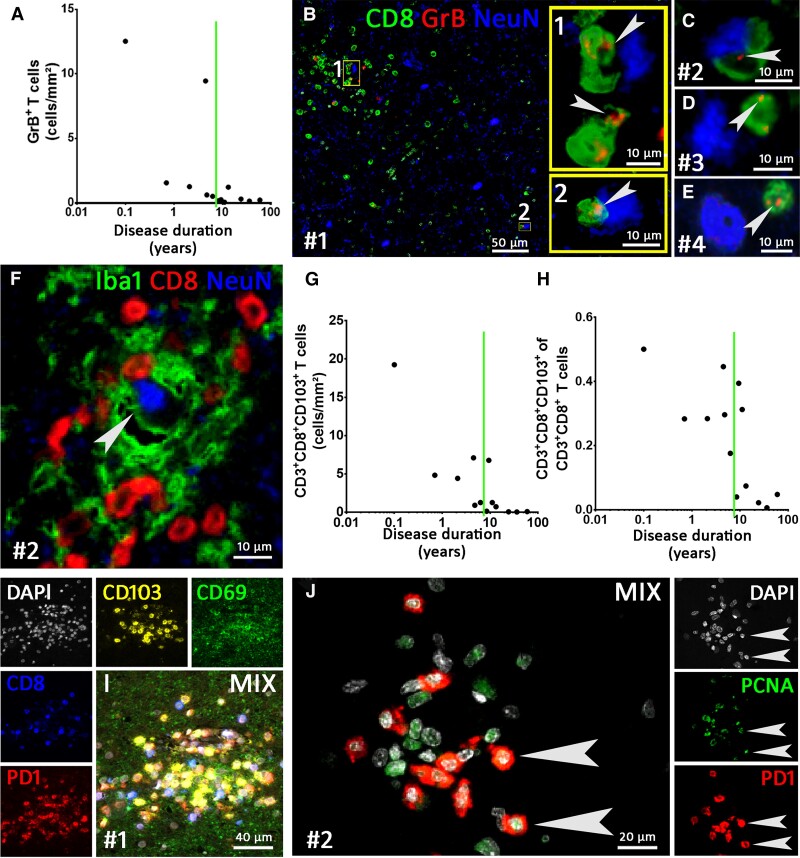
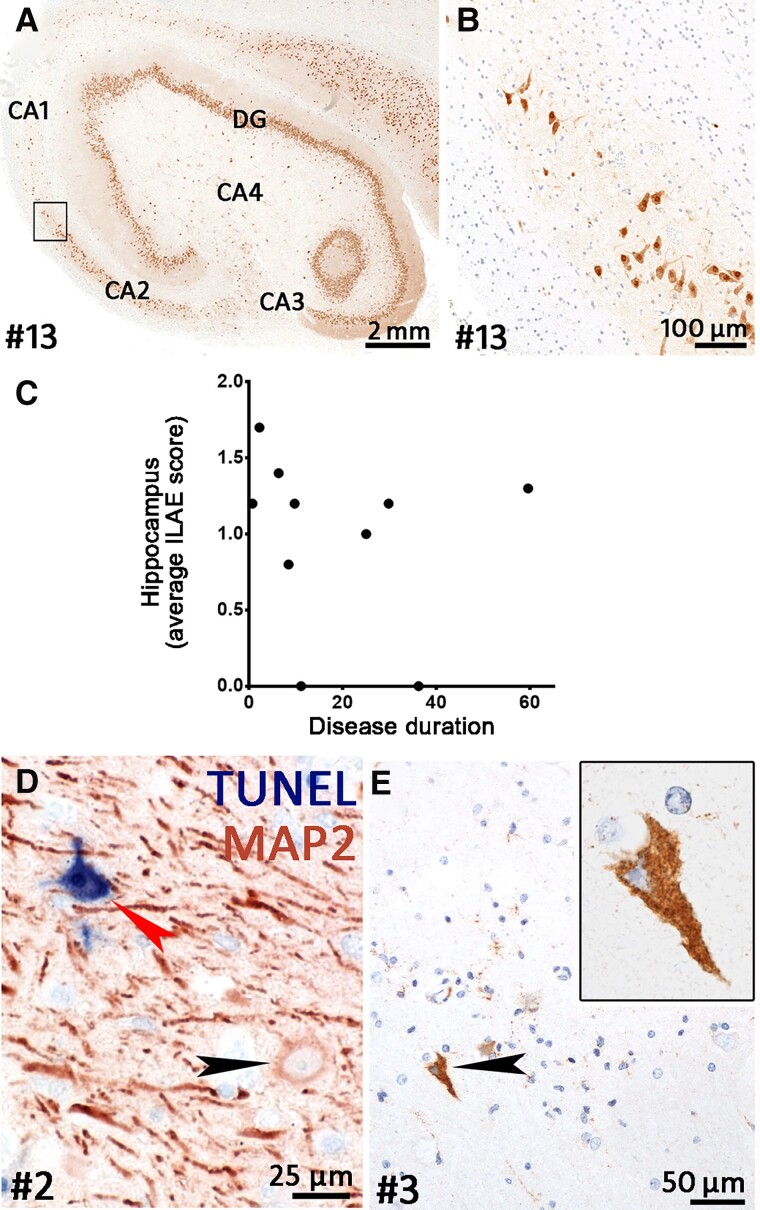
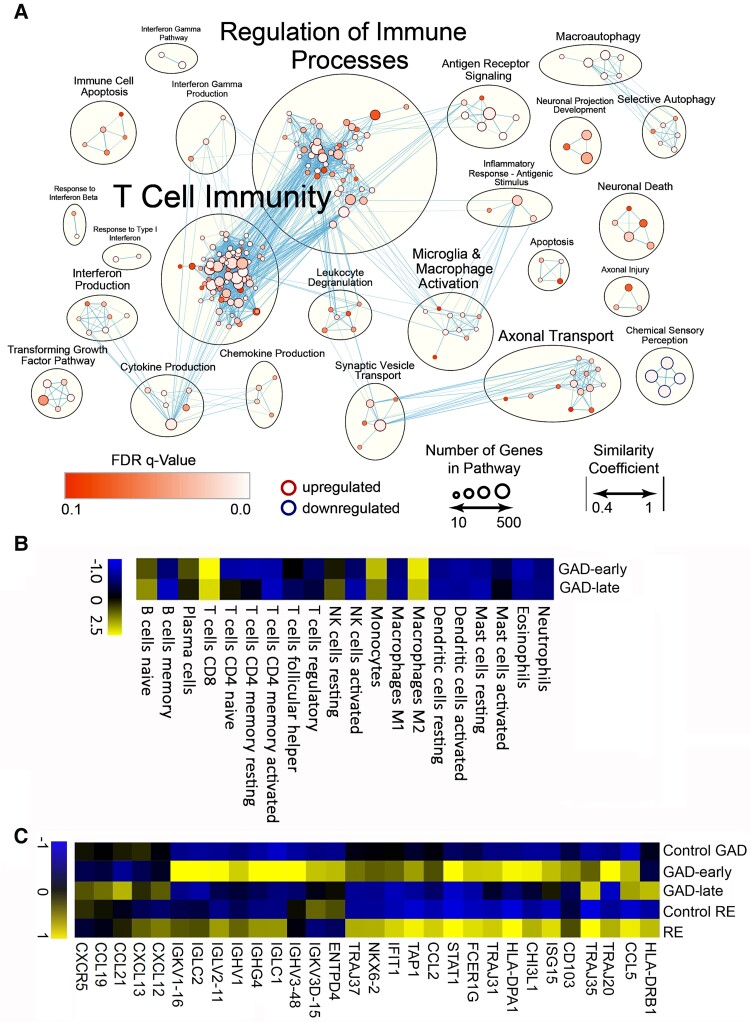
Temporal lobe epilepsy (TLE) is one of the syndromes linked to antibodies against glutamic acid decarboxylase (GAD). It has been questioned whether 'limbic encephalitis with GAD antibodies' is a meaningful diagnostic entity. The immunopathogenesis of GAD-TLE has remained enigmatic. Improvement of immunological treatability is an urgent clinical concern. We retrospectively assessed the clinical, MRI and CSF course as well as brain tissue of 15 adult patients with GAD-TLE who underwent temporal lobe surgery. Brain tissue was studied by means of immunohistochemistry, multiplex fluorescent microscopy and transcriptomic analysis for inflammatory mediators and neuronal degeneration. In 10 patients, there was a period of mediotemporal swelling and T2 signal increase; in nine cases this occurred within the first 6 years after symptom onset. This resulted in unilateral or bilateral hippocampal sclerosis; three cases developed hippocampal sclerosis within the first 2 years. All CSF studies done within the first year (n = 6) revealed intrathecal synthesis of immunoglobulin G. Temporal lobe surgeries were done after a median disease duration of 9 years (range 3 weeks to 60 years). Only two patients became seizure-free. Brain parenchyma collected during surgery in the first 6 years revealed high numbers of plasma cells but no signs of antibody-mediated tissue damage. Even more dense was the infiltration by CD8+ cytotoxic T lymphocytes (CTLs) that were seen to locally proliferate. Further, a portion of these cells revealed an antigen-specific resident memory T cell phenotype. Finally, CTLs with cytotoxic granzyme B+ granules were also seen in microglial nodules and attached to neurons, suggesting a CTL-mediated destruction of these cells. With longer disease duration, the density of all lymphocytes decreased. Whole transcriptome analysis in early/active cases (but not in late/inactive stages) revealed 'T cell immunity' and 'Regulation of immune processes' as the largest overrepresented clusters. To a lesser extent, pathways associated with B cells and neuronal degeneration also showed increased representation. Surgically treated patients with GAD-TLE go through an early active inflammatory, 'encephalitic' stage (≤6 years) with CTL-mediated, antigen-driven neuronal loss and antibody-producing plasma cells but without signs of complement-mediated cell death. Subsequently, patients enter an apparently immunologically inactive or low-active stage with ongoing seizures, probably caused by the structural damage to the temporal lobe. 'Limbic encephalitis' with GAD antibodies should be subsumed under GAD-TLE. The early tissue damage explains why immunotherapy does not usually lead to freedom from seizures.
Keywords: B cells; T cells; glutamic acid decarboxylase antibodies; histopathology; plasma cells; temporal lobe epilepsy.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
R.S. has received speaker fees or honoraria for serving on the advisory board from Angelini, Arvelle, Bial, Desitin, Eisai, LivaNova, UCB Pharma, UnEEG and Zogenix, unrelated to this work. B.V. was supported by travel expenses and fees by Livanova, Medtronic, UCB, EISAI, JAZZ and Bioprojet.
Figures
Comment in
-
Cytotoxic T cells and plasma cells dominate early in temporal lobe epilepsy with GAD antibodies.Brain. 2023 Apr 19;146(4):1231-1233. doi: 10.1093/brain/awad066. Brain. 2023. PMID: 36864690 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
