

pUL36 Deubiquitinase Activity Augments Both the Initiation and the Progression of Lytic Herpes Simplex Virus Infection in IFN-Primed Cells
- PMID: 36314822
- PMCID: PMC9683058
- DOI: 10.1128/jvi.00963-22
pUL36 Deubiquitinase Activity Augments Both the Initiation and the Progression of Lytic Herpes Simplex Virus Infection in IFN-Primed Cells
Abstract
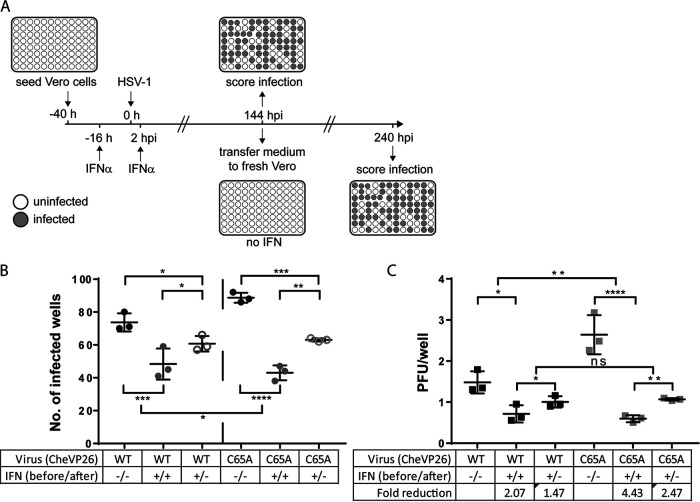
The evolutionarily conserved, structural HSV-1 tegument protein pUL36 is essential for both virus entry and assembly. While its N-terminal deubiquitinase (DUB) activity is dispensable for infection in cell culture, it is required for efficient virus spread in vivo, as it acts as a potent viral immune evasin. Interferon (IFN) induces the expression of hundreds of antiviral factors, including many ubiquitin modulators, which HSV-1 needs to neutralize to efficiently initiate a productive infection. Herein, we discover two functions of the conserved pUL36 DUB during lytic replication in cell culture in an understudied but equally important scenario of HSV-1 infection in IFN-treated cells. Our data indicate that the pUL36 DUB contributes to overcoming the IFN-mediated suppression of productive infection in both the early and late phases of HSV-1 infection. We show that incoming tegument-derived pUL36 DUB activity contributes to the IFN resistance of HSV-1 in IFN-primed cells to efficiently initiate lytic virus replication. Subsequently, the de novo expressed DUB augmented the efficiency of virus replication and increased the output of infectious virus. Notably, the DUB defect was only apparent when IFN was applied prior to infection. Our data indicate that IFN-induced defense mechanisms exist and that they work to both neutralize infectivity early on and slow the progression of HSV-1 replication in the late stages of infection. Also, our data indicate that pUL36 DUB activity contributes to the disarming of these host responses. IMPORTANCE HSV-1 is a ubiquitous human pathogen that is responsible for common cold sores and may also cause life-threatening disease. pUL36 is an essential, conserved herpesvirus protein with N-terminal deubiquitinating (DUB) activity. The DUB is dispensable for HSV-1 replication in cell culture but represents an important viral immune evasin in vivo. IFN plays a pivotal role in HSV-1 infection and suppresses viral replication both in vitro and in vivo. Here, we show that DUB activity contributes to overcoming IFN-induced cellular resistance in order to more efficiently initiate lytic replication and produce infectious virions. As such, DUB activity in the incoming virions increases their infectivity, while the de novo synthesized DUB augments productive infection. Thus, the HSV-1 DUB antagonizes the activity of IFN-inducible effector proteins to facilitate productive infection at multiple levels. Our findings underscore the importance of using more challenging cell culture systems to fully understand virus protein functions.
Keywords: DUB; HSV-1; UL36; USP; herpes simplex virus 1; innate immunity; interferon; interferon antagonism; ubiquitin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





Similar articles
-
Conserved Tryptophan Motifs in the Large Tegument Protein pUL36 Are Required for Efficient Secondary Envelopment of Herpes Simplex Virus Capsids.J Virol. 2016 May 12;90(11):5368-5383. doi: 10.1128/JVI.03167-15. Print 2016 Jun 1. J Virol. 2016. PMID: 27009950 Free PMC article.
-
The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus.J Virol. 2012 Apr;86(7):3682-700. doi: 10.1128/JVI.06432-11. Epub 2012 Jan 18. J Virol. 2012. PMID: 22258258 Free PMC article.
-
Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-κB Activation in DNA Sensing Signal Pathway.J Virol. 2017 Feb 14;91(5):e02417-16. doi: 10.1128/JVI.02417-16. Print 2017 Mar 1. J Virol. 2017. PMID: 28031360 Free PMC article.
-
HSV Replication: Triggering and Repressing STING Functionality.Viruses. 2023 Jan 13;15(1):226. doi: 10.3390/v15010226. Viruses. 2023. PMID: 36680267 Free PMC article. Review.
-
Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms.Front Immunol. 2021 May 31;12:644664. doi: 10.3389/fimmu.2021.644664. eCollection 2021. Front Immunol. 2021. PMID: 34135889 Free PMC article. Review.
Cited by
-
The precise function of alphaherpesvirus tegument proteins and their interactions during the viral life cycle.Front Microbiol. 2024 Jul 2;15:1431672. doi: 10.3389/fmicb.2024.1431672. eCollection 2024. Front Microbiol. 2024. PMID: 39015737 Free PMC article. Review.
-
The TET3 inflammasome senses unique long HSV-1 proteins for virus particle budding from the nucleus.Cell Mol Immunol. 2024 Nov;21(11):1322-1334. doi: 10.1038/s41423-024-01221-2. Epub 2024 Oct 8. Cell Mol Immunol. 2024. PMID: 39379602
-
Tethered release of the pseudorabies virus deubiquitinase from the capsid promotes enzymatic activity.J Virol. 2025 Jan 31;99(1):e0151724. doi: 10.1128/jvi.01517-24. Epub 2024 Dec 5. J Virol. 2025. PMID: 39636112 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
