

Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia
- PMID: 36319618
- PMCID: PMC9626559
- DOI: 10.1038/s41467-022-33395-6
Gut microbiome dysbiosis in antibiotic-treated COVID-19 patients is associated with microbial translocation and bacteremia
Abstract
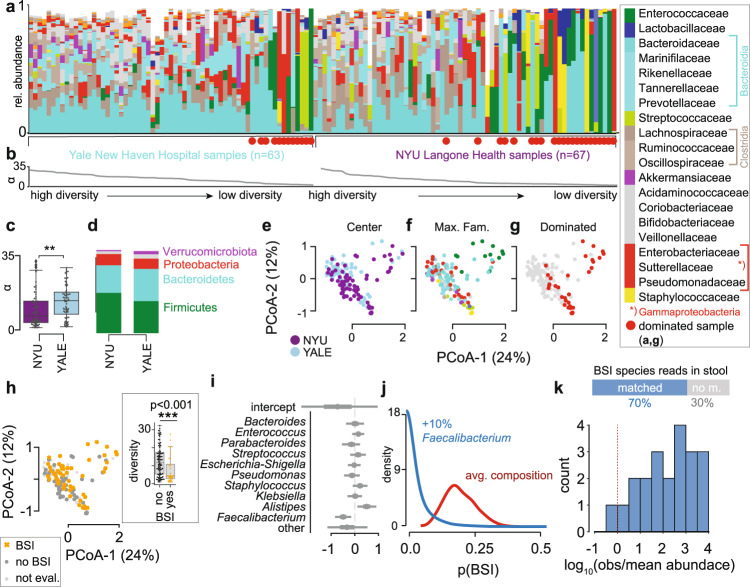
Although microbial populations in the gut microbiome are associated with COVID-19 severity, a causal impact on patient health has not been established. Here we provide evidence that gut microbiome dysbiosis is associated with translocation of bacteria into the blood during COVID-19, causing life-threatening secondary infections. We first demonstrate SARS-CoV-2 infection induces gut microbiome dysbiosis in mice, which correlated with alterations to Paneth cells and goblet cells, and markers of barrier permeability. Samples collected from 96 COVID-19 patients at two different clinical sites also revealed substantial gut microbiome dysbiosis, including blooms of opportunistic pathogenic bacterial genera known to include antimicrobial-resistant species. Analysis of blood culture results testing for secondary microbial bloodstream infections with paired microbiome data indicates that bacteria may translocate from the gut into the systemic circulation of COVID-19 patients. These results are consistent with a direct role for gut microbiome dysbiosis in enabling dangerous secondary infections during COVID-19.
© 2022. The Author(s).
Conflict of interest statement
K.C. has received research support from Pfizer, Takeda, Pacific Biosciences, Genentech, and Abbvie; consulted for or received an honoraria from Puretech Health, Genentech, and Abbvie; and is named as an inventor on U.S. patent 10,722,600 and provisional patents 62/935,035 and 63/157,225. J.S. is cofounder of Postbiotics Plus Research LLC. The remaining authors declare no competing interests.
Figures


Update of
-
Gut microbiome dysbiosis during COVID-19 is associated with increased risk for bacteremia and microbial translocation.Res Sq [Preprint]. 2021 Jul 27:rs.3.rs-726620. doi: 10.21203/rs.3.rs-726620/v1. Res Sq. 2021. Update in: Nat Commun. 2022 Nov 1;13(1):5926. doi: 10.1038/s41467-022-33395-6. PMID: 34341786 Free PMC article. Updated. Preprint.
-
Gut microbiome dysbiosis during COVID-19 is associated with increased risk for bacteremia and microbial translocation.bioRxiv [Preprint]. 2022 Mar 2:2021.07.15.452246. doi: 10.1101/2021.07.15.452246. bioRxiv. 2022. Update in: Nat Commun. 2022 Nov 1;13(1):5926. doi: 10.1038/s41467-022-33395-6. PMID: 35262080 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- K23 DK124570/DK/NIDDK NIH HHS/United States
- P30 CA016087/CA/NCI NIH HHS/United States
- R01 DK093668/DK/NIDDK NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- R21 AI139374/AI/NIAID NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- OT2 HL161847/HL/NHLBI NIH HHS/United States
- R01 AI140754/AI/NIAID NIH HHS/United States
- S10 OD021747/OD/NIH HHS/United States
- R01 AI121244/AI/NIAID NIH HHS/United States
- T32 AI100853/AI/NIAID NIH HHS/United States
- R01 AI143639/AI/NIAID NIH HHS/United States
- R01 HL123340/HL/NHLBI NIH HHS/United States
- R01 AI130945/AI/NIAID NIH HHS/United States
- R01 CA269617/CA/NCI NIH HHS/United States
- DP2 AI164318/AI/NIAID NIH HHS/United States
- R01 DK124336/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
