MIDAS2: Metagenomic Intra-species Diversity Analysis System
- PMID: 36321886
- PMCID: PMC9805558
- DOI: 10.1093/bioinformatics/btac713
MIDAS2: Metagenomic Intra-species Diversity Analysis System
Abstract
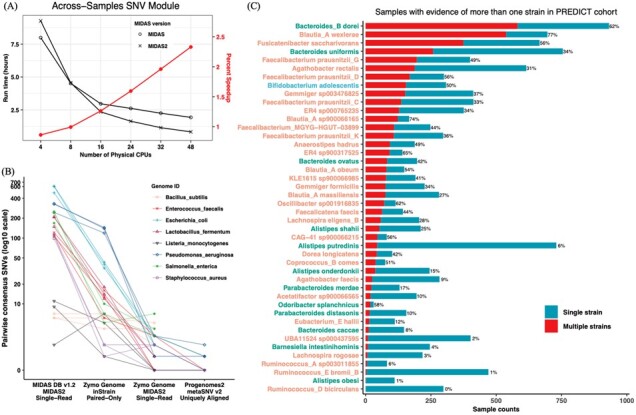
Summary: The Metagenomic Intra-Species Diversity Analysis System (MIDAS) is a scalable metagenomic pipeline that identifies single nucleotide variants (SNVs) and gene copy number variants in microbial populations. Here, we present MIDAS2, which addresses the computational challenges presented by increasingly large reference genome databases, while adding functionality for building custom databases and leveraging paired-end reads to improve SNV accuracy. This fast and scalable reengineering of the MIDAS pipeline enables thousands of metagenomic samples to be efficiently genotyped.
Availability and implementation: The source code is available at https://github.com/czbiohub/MIDAS2.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures