

C1Q labels a highly aggressive macrophage-like leukemia population indicating extramedullary infiltration and relapse
- PMID: 36322939
- PMCID: PMC10651790
- DOI: 10.1182/blood.2022017046
C1Q labels a highly aggressive macrophage-like leukemia population indicating extramedullary infiltration and relapse
Abstract
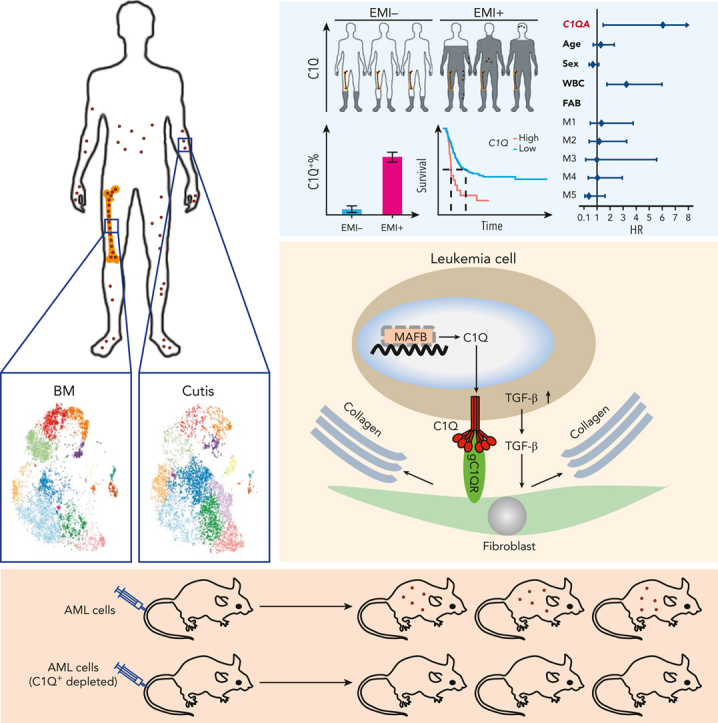
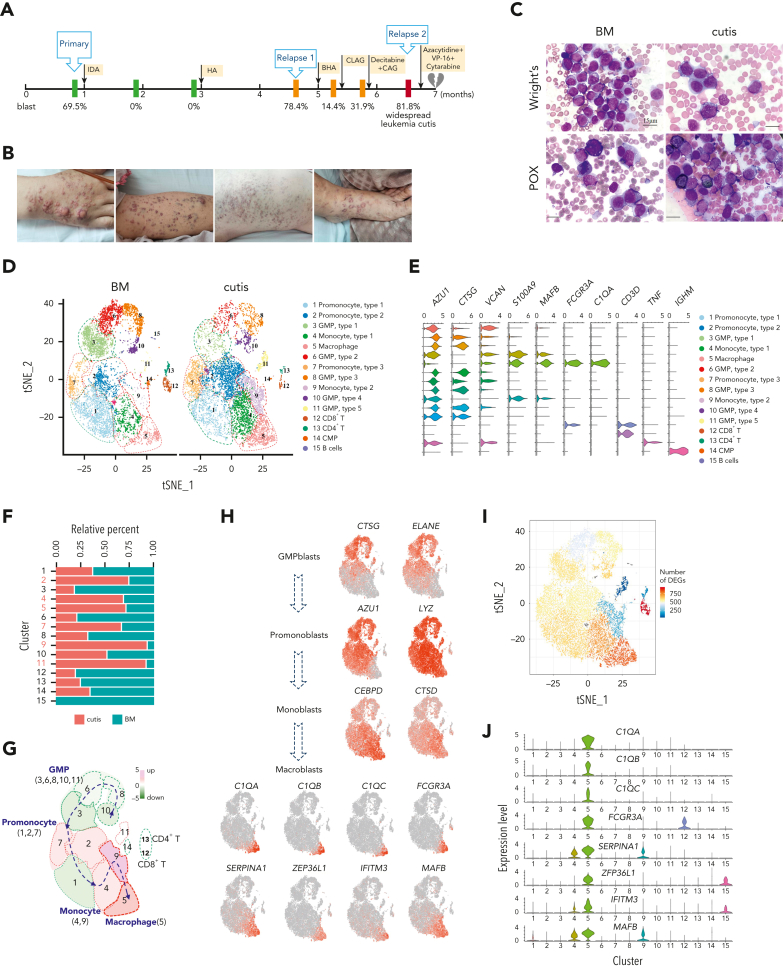
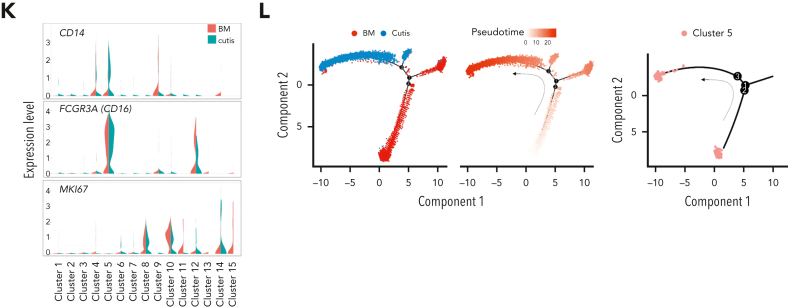
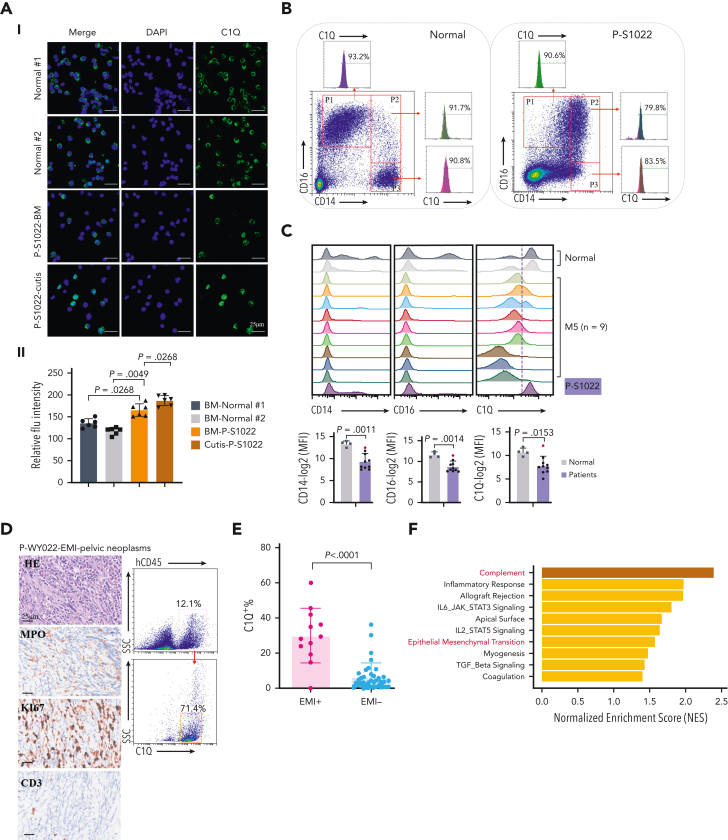
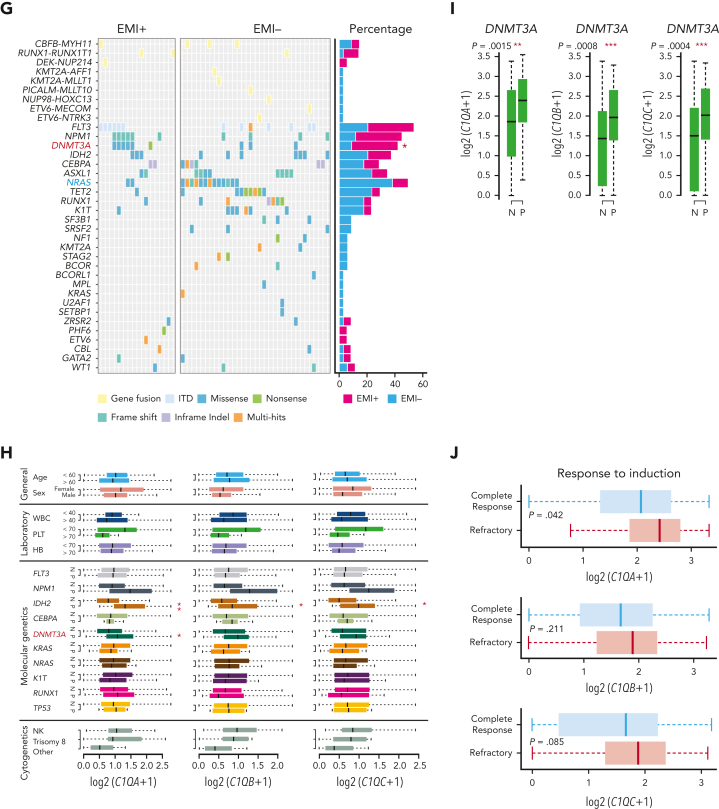
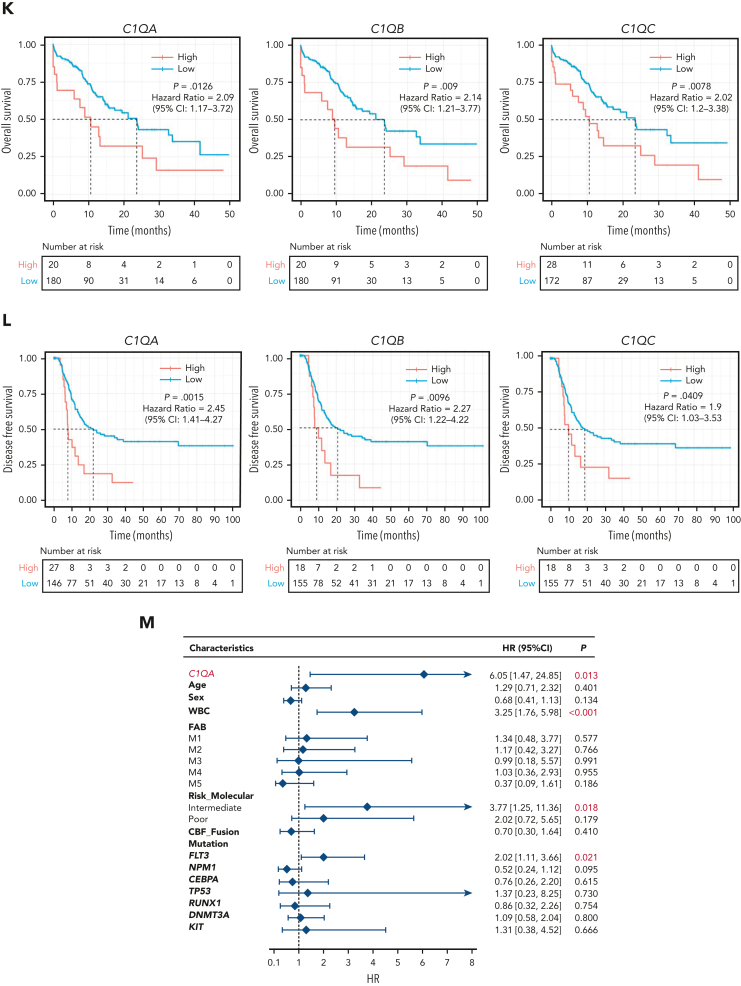
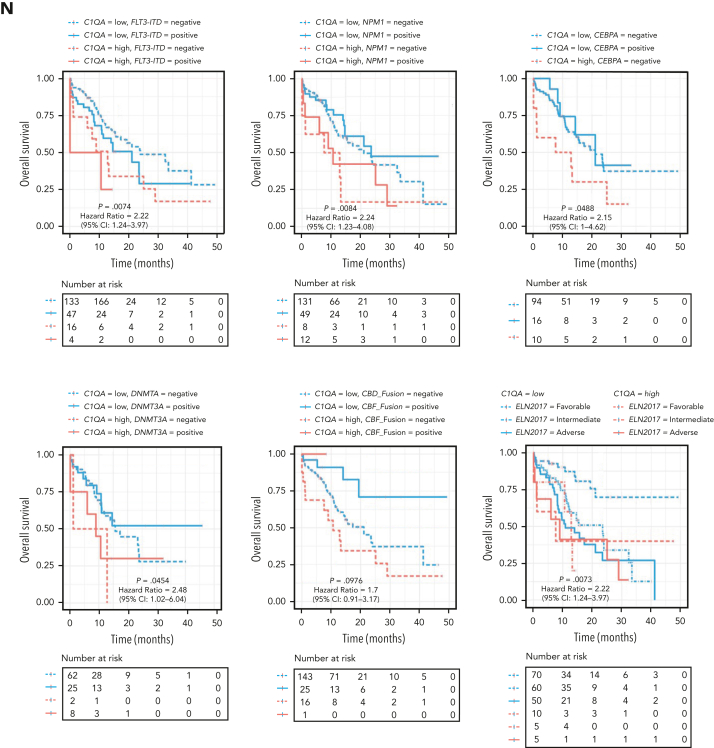
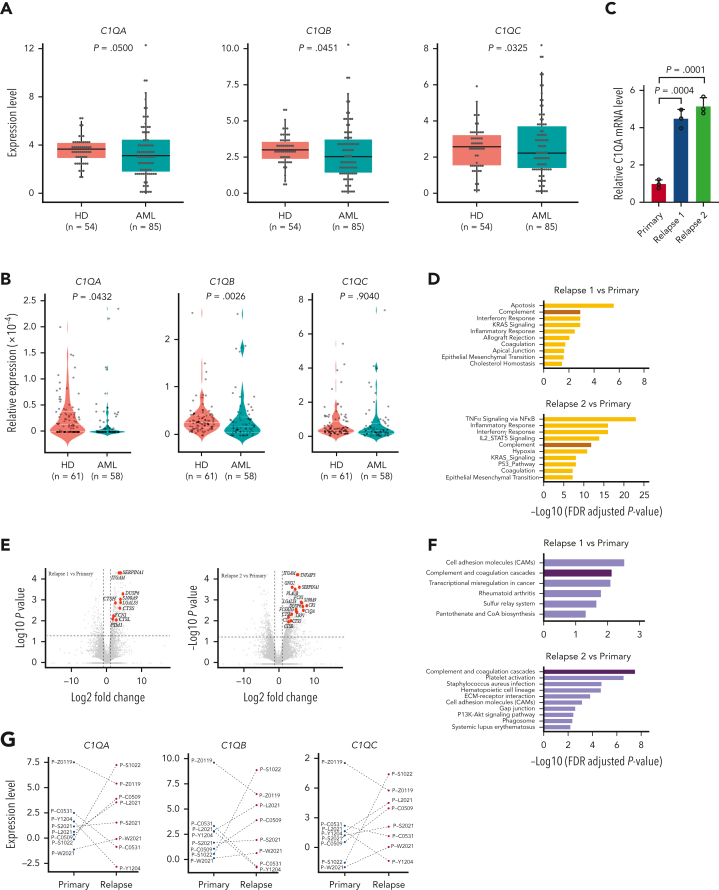
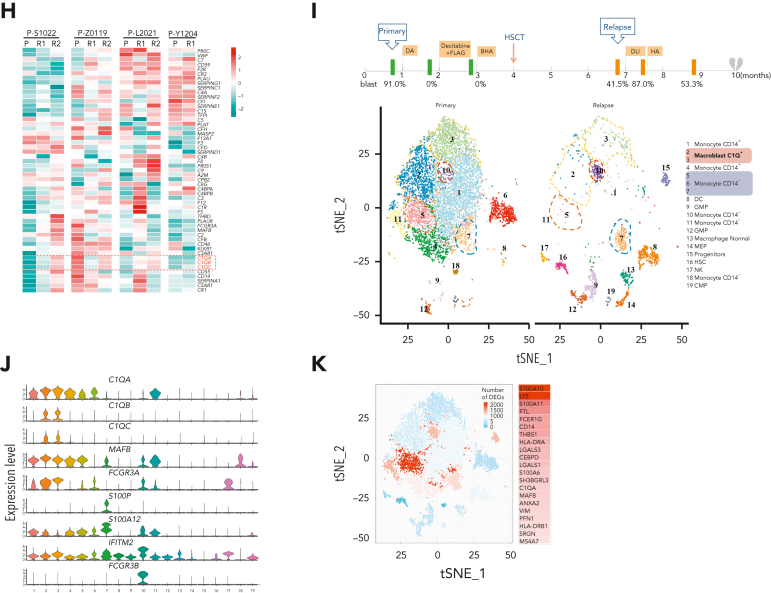
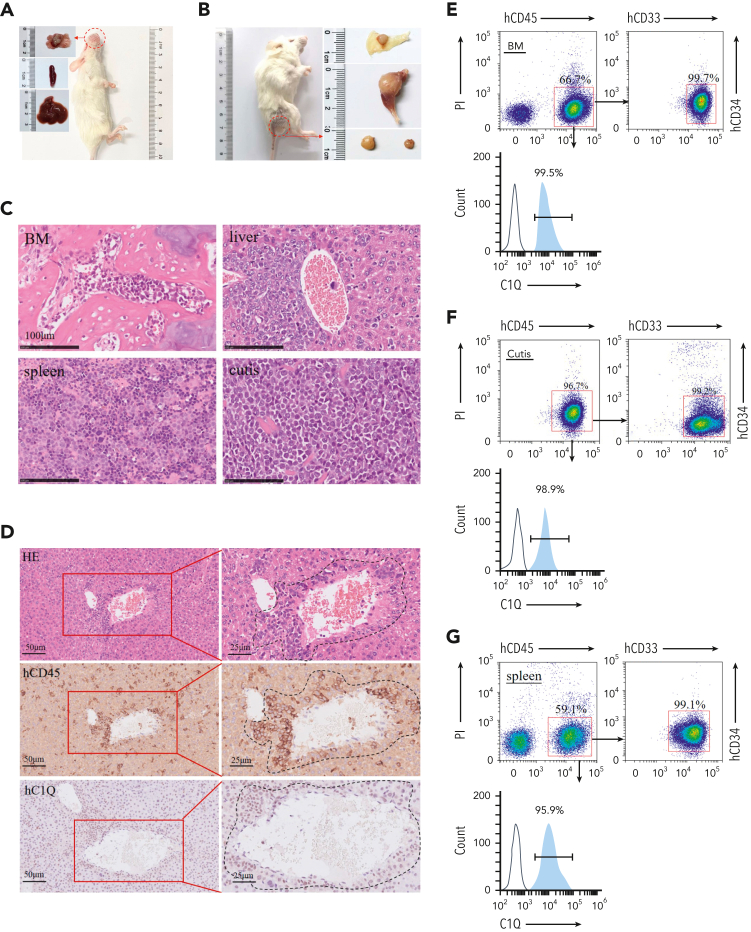
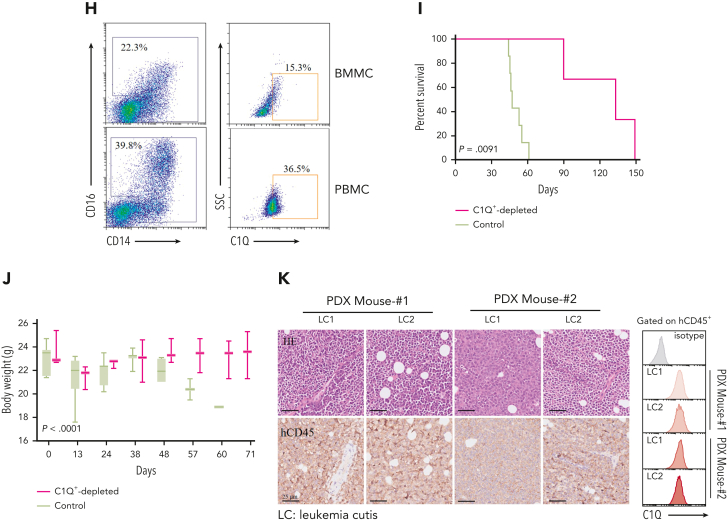
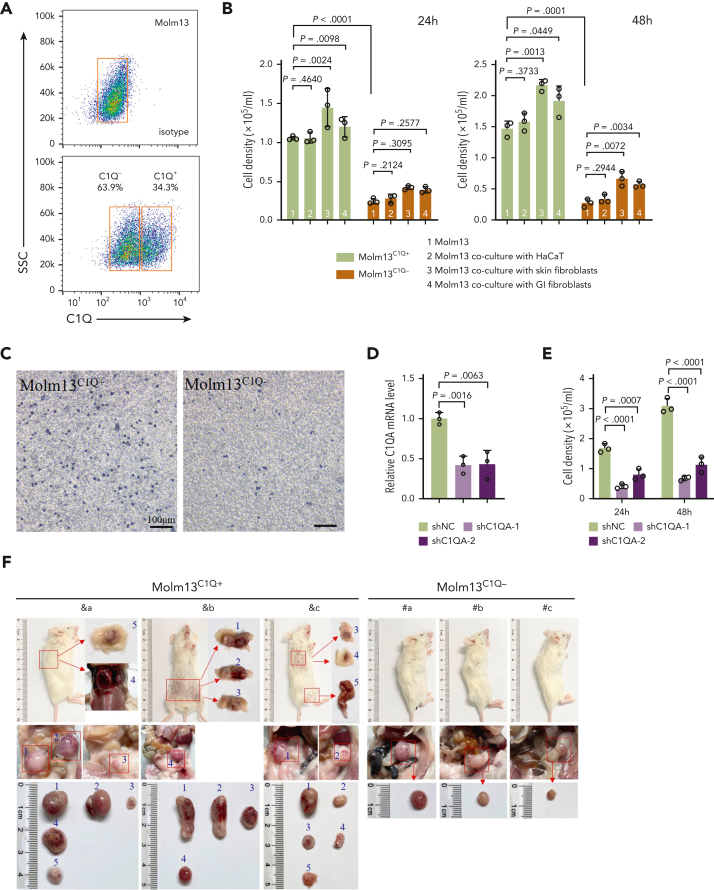
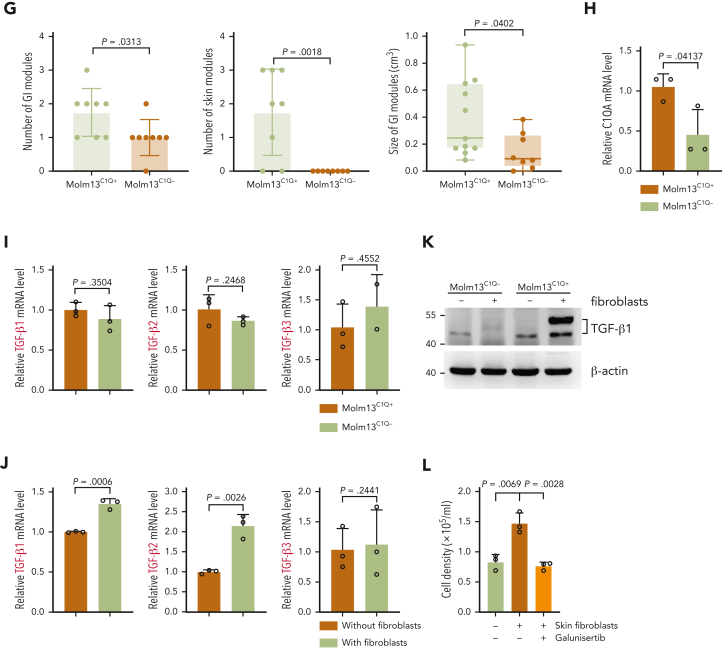
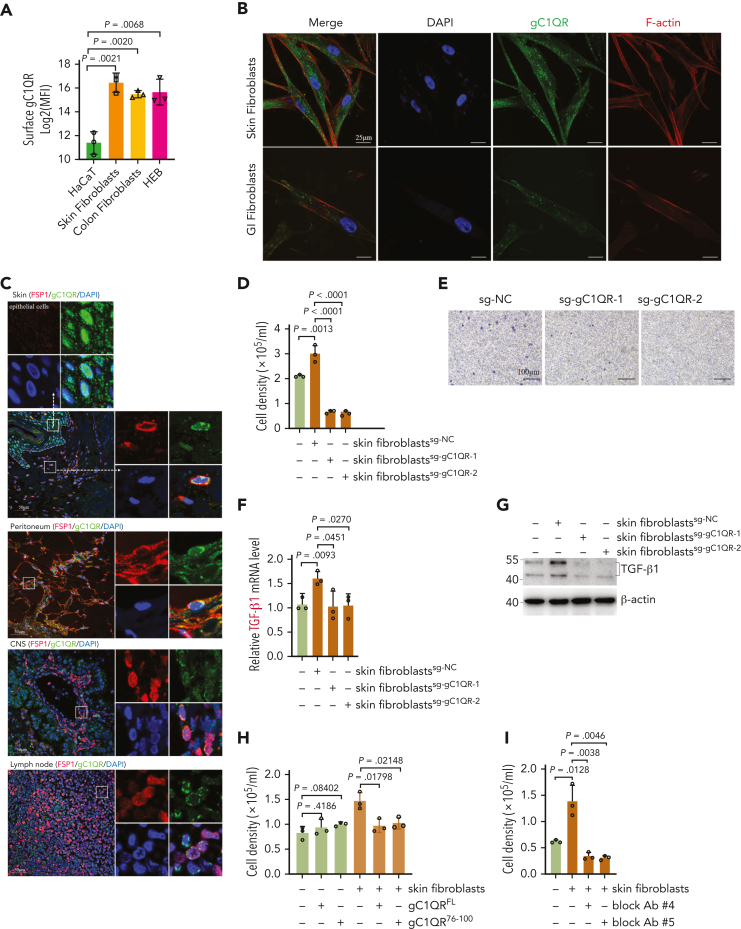
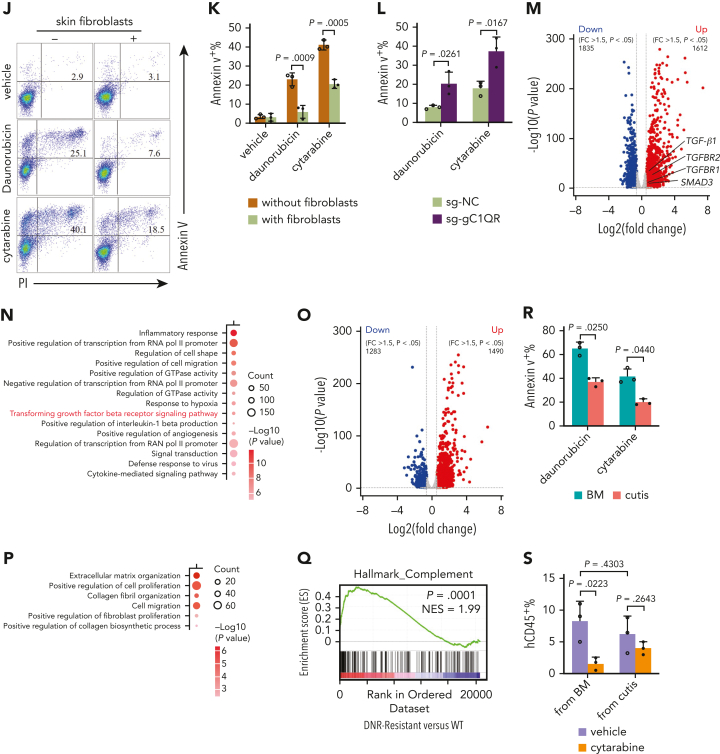
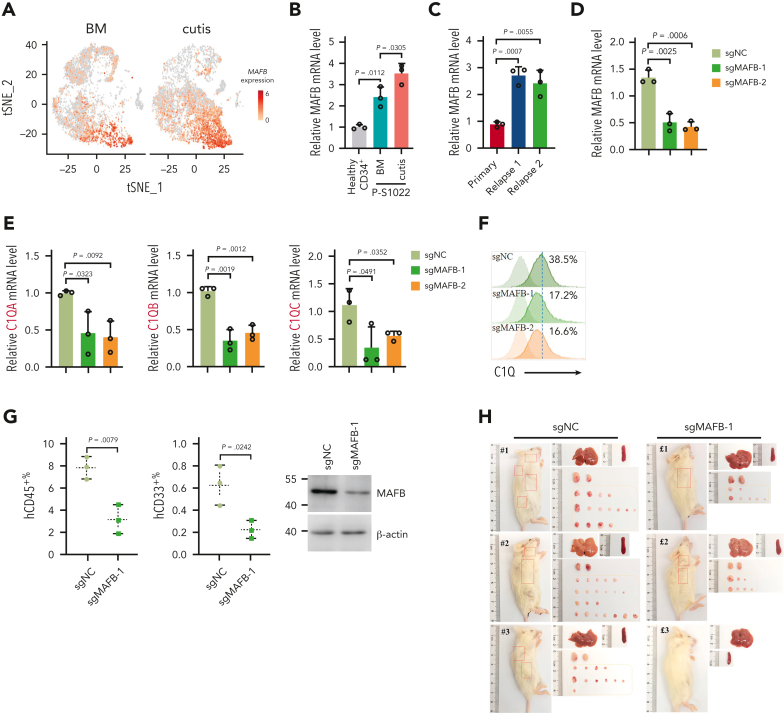
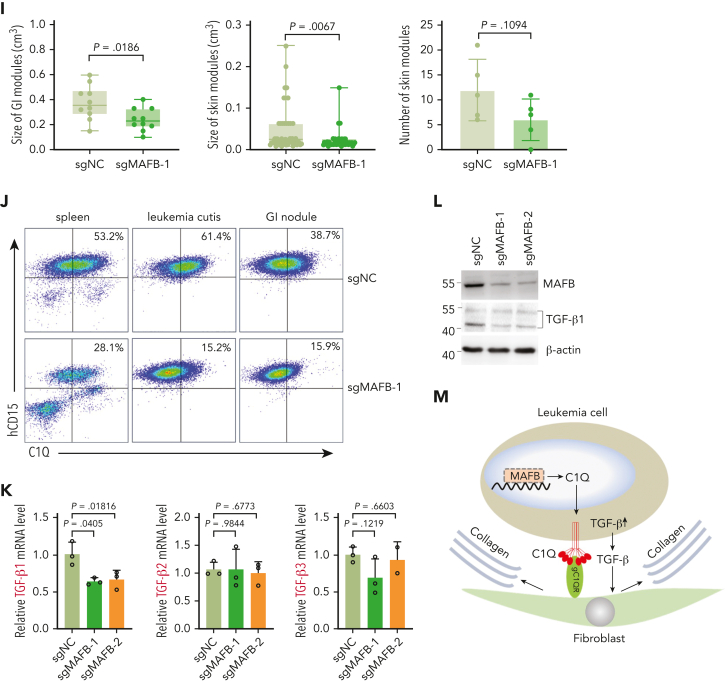
Extramedullary infiltration (EMI) is a concomitant manifestation that may indicate poor outcome of acute myeloid leukemia (AML). The underlying mechanism remains poorly understood and therapeutic options are limited. Here, we employed single-cell RNA sequencing on bone marrow (BM) and EMI samples from a patient with AML presenting pervasive leukemia cutis. A complement C1Q+ macrophage-like leukemia subset, which was enriched within cutis and existed in BM before EMI manifestations, was identified and further verified in multiple patients with AML. Genomic and transcriptional profiling disclosed mutation and gene expression signatures of patients with EMI that expressed high levels of C1Q. RNA sequencing and quantitative proteomic analysis revealed expression dynamics of C1Q from primary to relapse. Univariate and multivariate analysis demonstrated adverse prognosis significance of C1Q expression. Mechanistically, C1Q expression, which was modulated by transcription factor MAF BZIP transcription factor B, endowed leukemia cells with tissue infiltration ability, which could establish prominent cutaneous or gastrointestinal EMI nodules in patient-derived xenograft and cell line-derived xenograft models. Fibroblasts attracted migration of the C1Q+ leukemia cells through C1Q-globular C1Q receptor recognition and subsequent stimulation of transforming growth factor β1. This cell-to-cell communication also contributed to survival of C1Q+ leukemia cells under chemotherapy stress. Thus, C1Q served as a marker for AML with adverse prognosis, orchestrating cancer infiltration pathways through communicating with fibroblasts and represents a compelling therapeutic target for EMI.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
C1q helps AML to disseminate and resist.Blood. 2023 Feb 16;141(7):691-692. doi: 10.1182/blood.2022018785. Blood. 2023. PMID: 36795452 No abstract available.
References
-
- Martin-Subero JI. Predicting leukemia relapse. Nat Med. 2018;24(4):385–387. - PubMed
-
- Marchesi V. Genetics: the AML mutational landscape. Nat Rev Clin Oncol. 2013;10(6):305. - PubMed
-
- Shahin OA, Ravandi F. Myeloid sarcoma. Curr Opin Hematol. 2020;27(2):88–94. - PubMed
-
- Bakst RL, Tallman MS, Douer D, Yahalom J. How I treat extramedullary acute myeloid leukemia. Blood. 2011;118(14):3785–3793. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
