

Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD
- PMID: 36323788
- PMCID: PMC9668748
- DOI: 10.1038/s41586-022-05377-7
Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD
Abstract
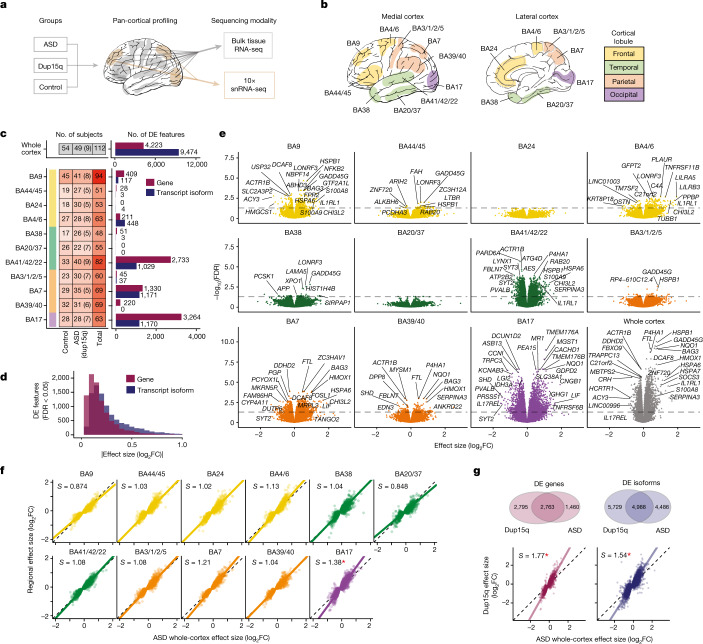
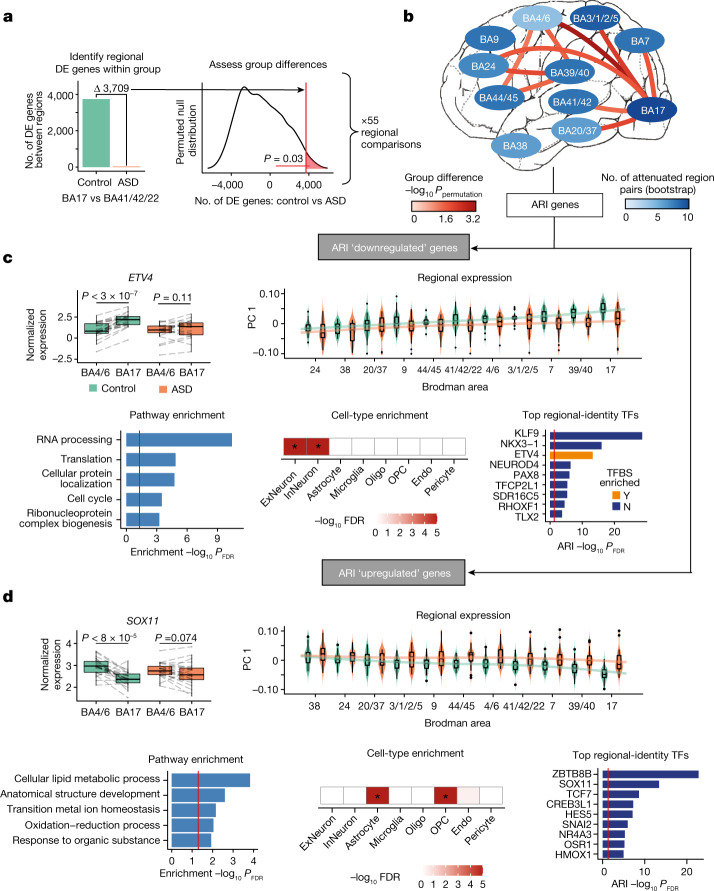
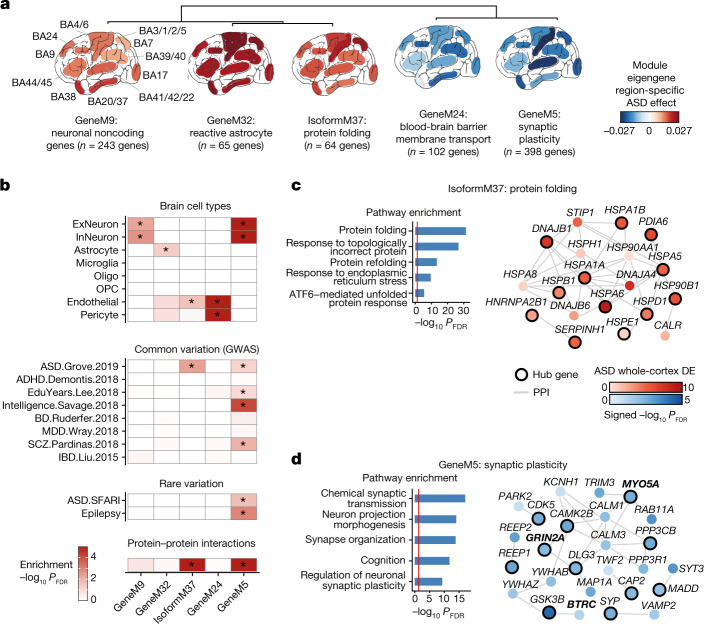
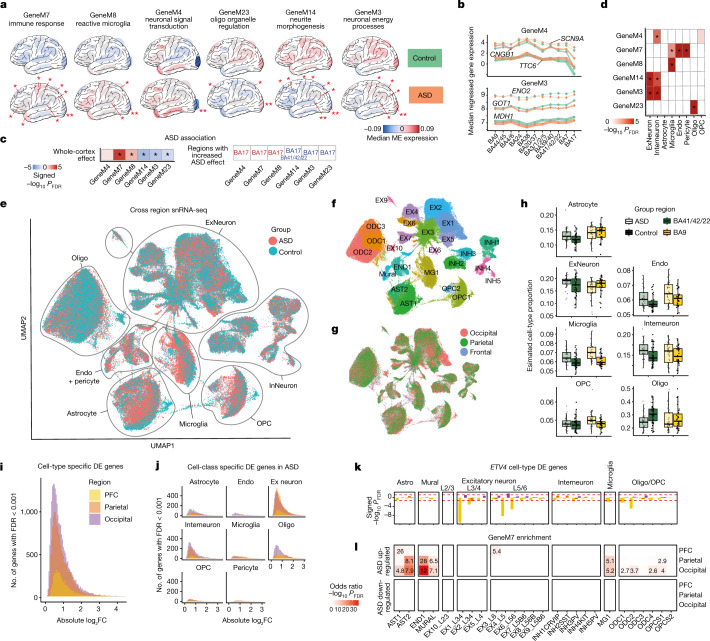
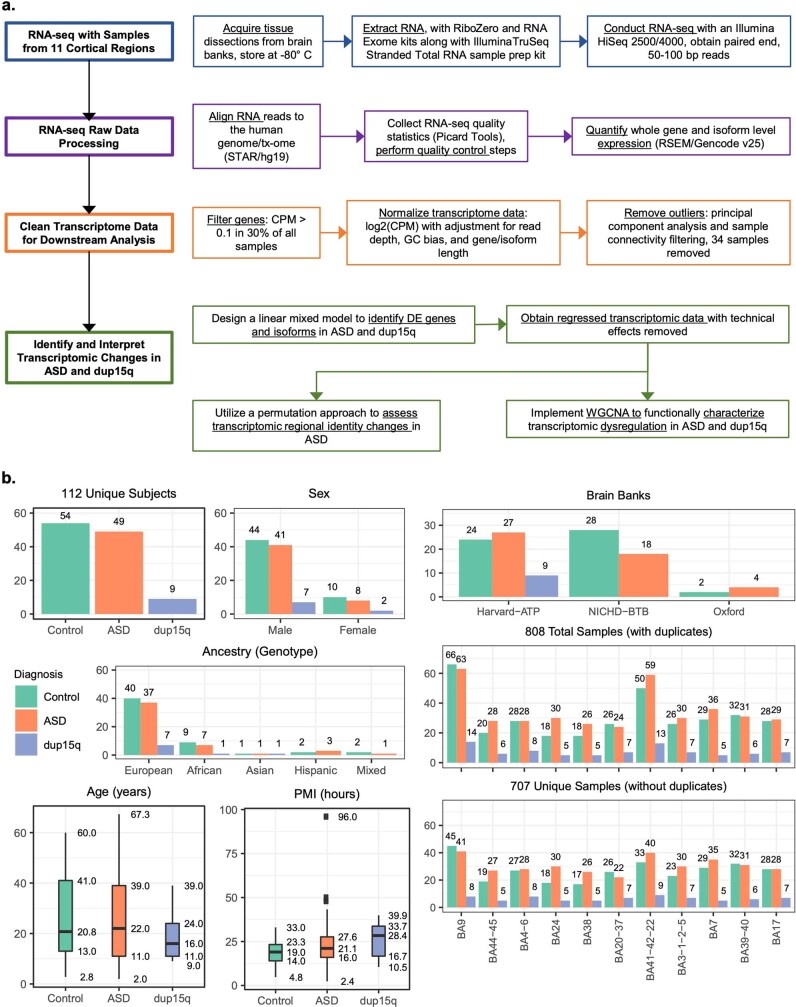
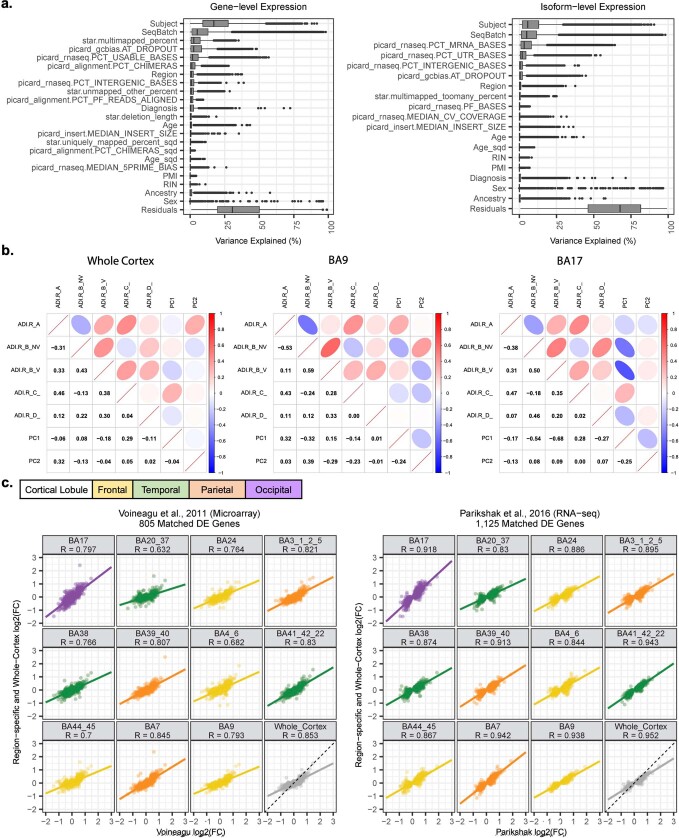
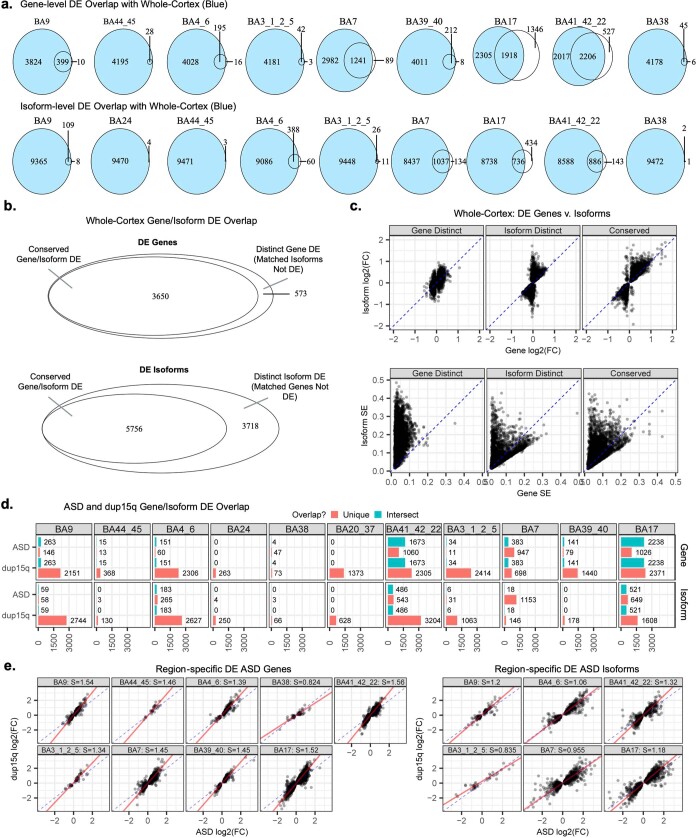
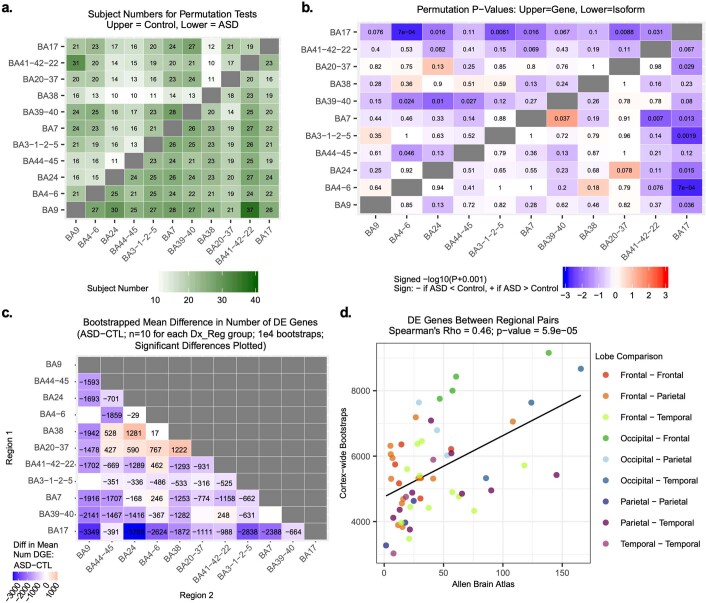
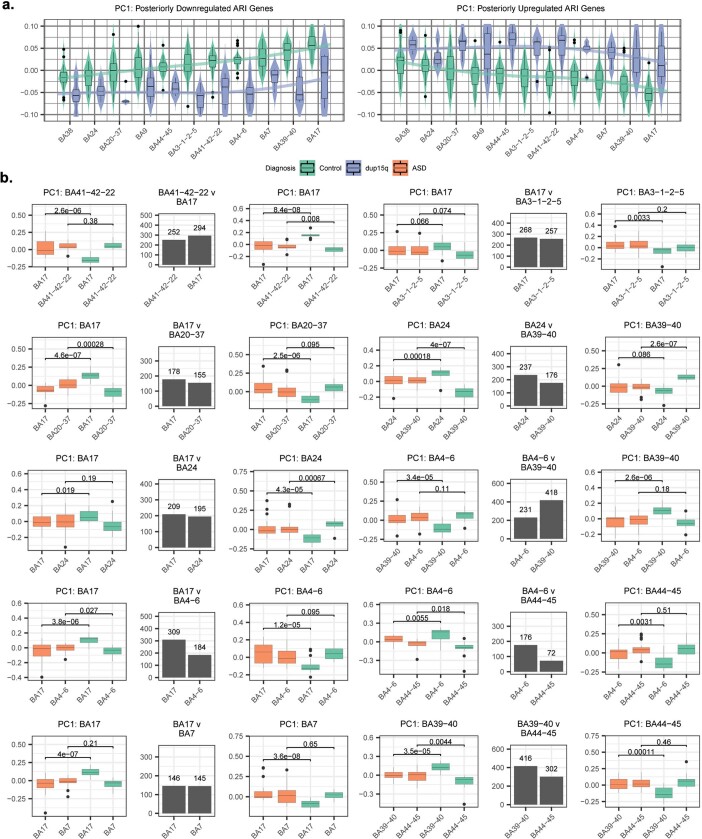
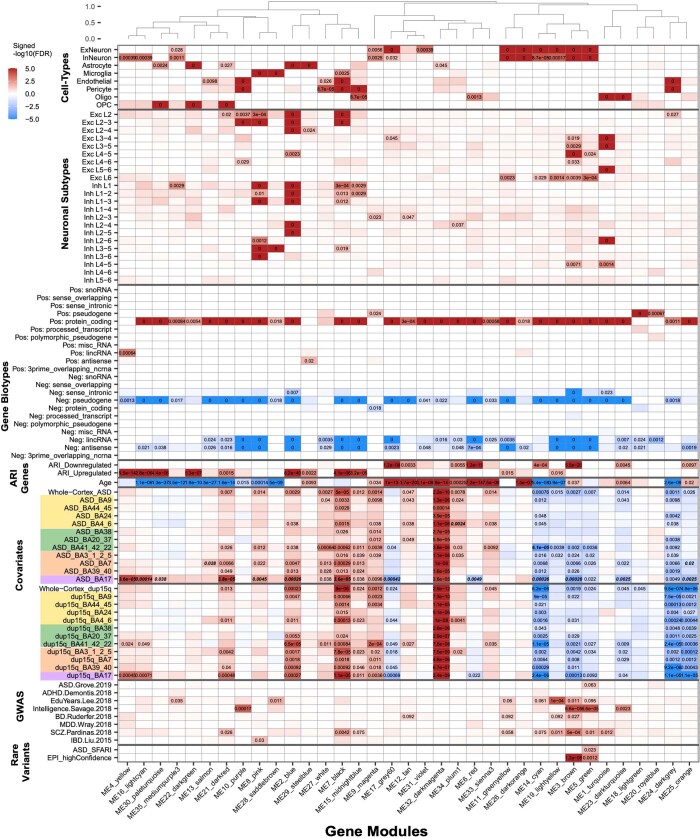
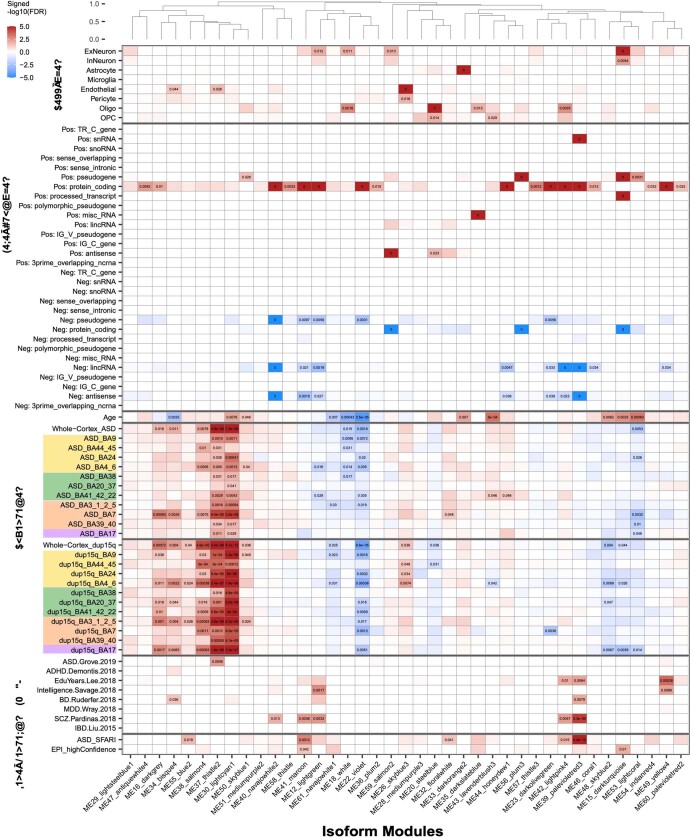
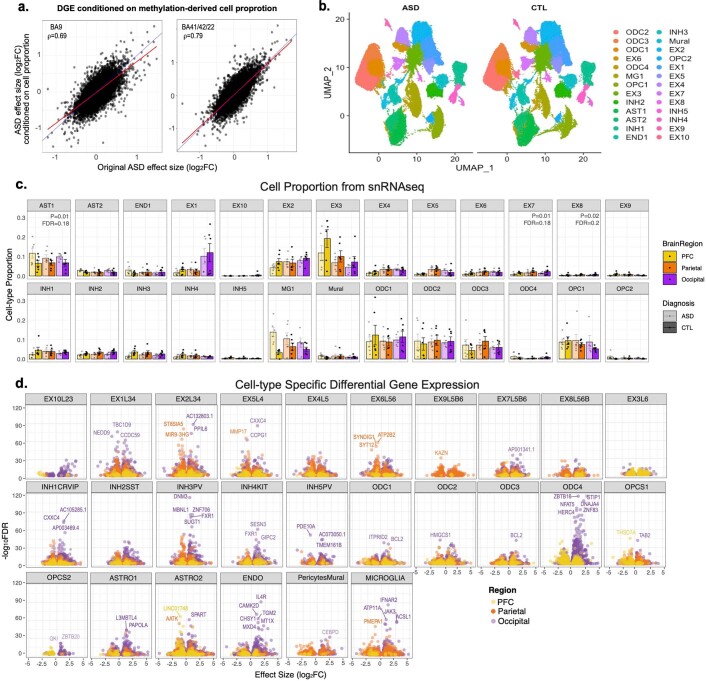
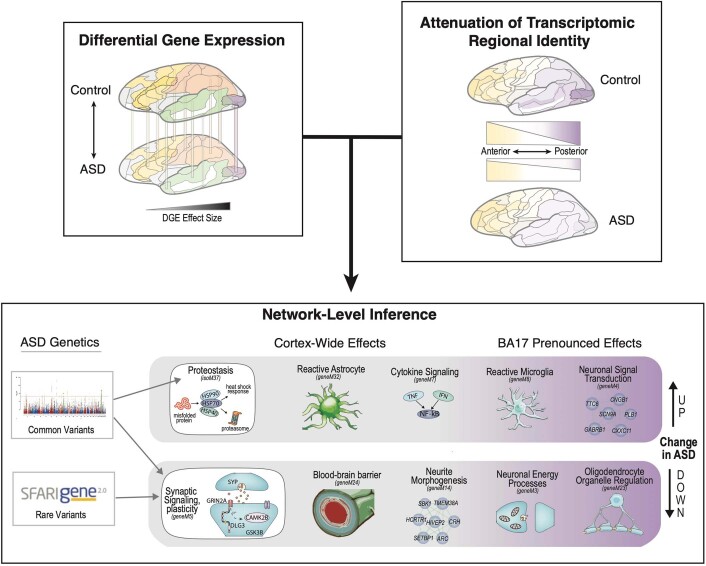
Neuropsychiatric disorders classically lack defining brain pathologies, but recent work has demonstrated dysregulation at the molecular level, characterized by transcriptomic and epigenetic alterations1-3. In autism spectrum disorder (ASD), this molecular pathology involves the upregulation of microglial, astrocyte and neural-immune genes, the downregulation of synaptic genes, and attenuation of gene-expression gradients in cortex1,2,4-6. However, whether these changes are limited to cortical association regions or are more widespread remains unknown. To address this issue, we performed RNA-sequencing analysis of 725 brain samples spanning 11 cortical areas from 112 post-mortem samples from individuals with ASD and neurotypical controls. We find widespread transcriptomic changes across the cortex in ASD, exhibiting an anterior-to-posterior gradient, with the greatest differences in primary visual cortex, coincident with an attenuation of the typical transcriptomic differences between cortical regions. Single-nucleus RNA-sequencing and methylation profiling demonstrate that this robust molecular signature reflects changes in cell-type-specific gene expression, particularly affecting excitatory neurons and glia. Both rare and common ASD-associated genetic variation converge within a downregulated co-expression module involving synaptic signalling, and common variation alone is enriched within a module of upregulated protein chaperone genes. These results highlight widespread molecular changes across the cerebral cortex in ASD, extending beyond association cortex to broadly involve primary sensory regions.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Sun W, et al. Histone acetylome-wide association study of autism spectrum disorder. Cell. 2016;167:1385–1397.e11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RF1 AG071683/AG/NIA NIH HHS/United States
- R01 MH125252/MH/NIMH NIH HHS/United States
- R01 MH094714/MH/NIMH NIH HHS/United States
- U01 MH115746/MH/NIMH NIH HHS/United States
- R01 MH109912/MH/NIMH NIH HHS/United States
- P50 MH106438/MH/NIMH NIH HHS/United States
- R01 AG071683/AG/NIA NIH HHS/United States
- P50 HD103557/HD/NICHD NIH HHS/United States
- F32 MH124337/MH/NIMH NIH HHS/United States
- R01 MH121521/MH/NIMH NIH HHS/United States
- R01 MH123922/MH/NIMH NIH HHS/United States
- R01 MH110927/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
