

Target-enriched long-read sequencing (TELSeq) contextualizes antimicrobial resistance genes in metagenomes
- PMID: 36324140
- PMCID: PMC9628182
- DOI: 10.1186/s40168-022-01368-y
Target-enriched long-read sequencing (TELSeq) contextualizes antimicrobial resistance genes in metagenomes
Abstract
Background: Metagenomic data can be used to profile high-importance genes within microbiomes. However, current metagenomic workflows produce data that suffer from low sensitivity and an inability to accurately reconstruct partial or full genomes, particularly those in low abundance. These limitations preclude colocalization analysis, i.e., characterizing the genomic context of genes and functions within a metagenomic sample. Genomic context is especially crucial for functions associated with horizontal gene transfer (HGT) via mobile genetic elements (MGEs), for example antimicrobial resistance (AMR). To overcome this current limitation of metagenomics, we present a method for comprehensive and accurate reconstruction of antimicrobial resistance genes (ARGs) and MGEs from metagenomic DNA, termed target-enriched long-read sequencing (TELSeq).
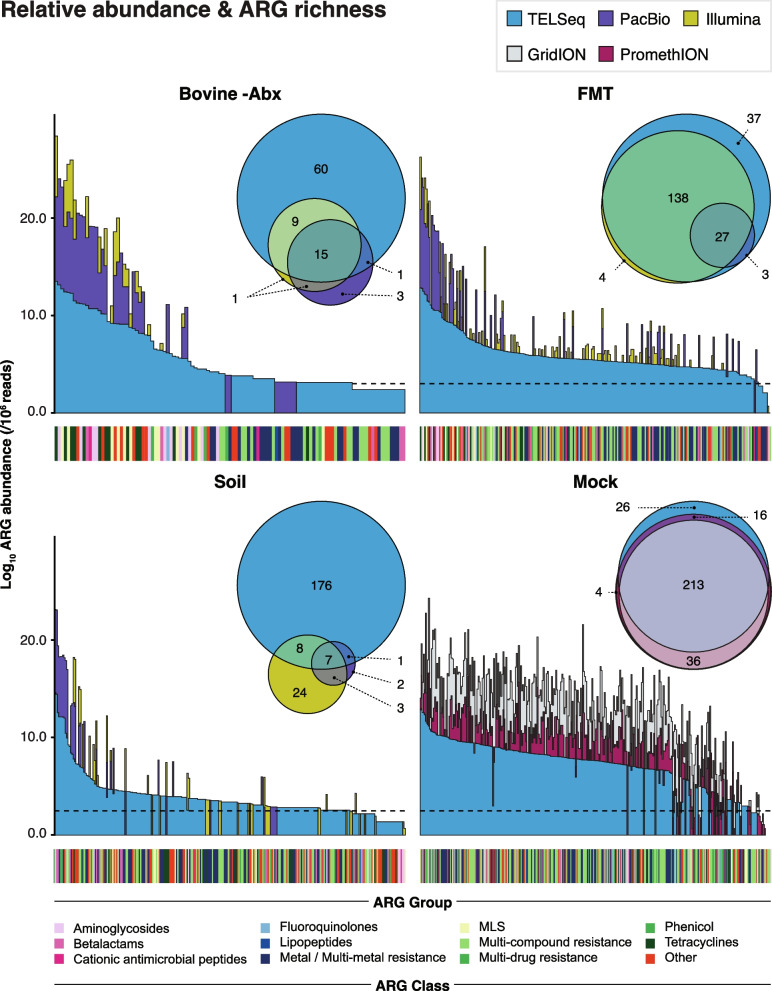
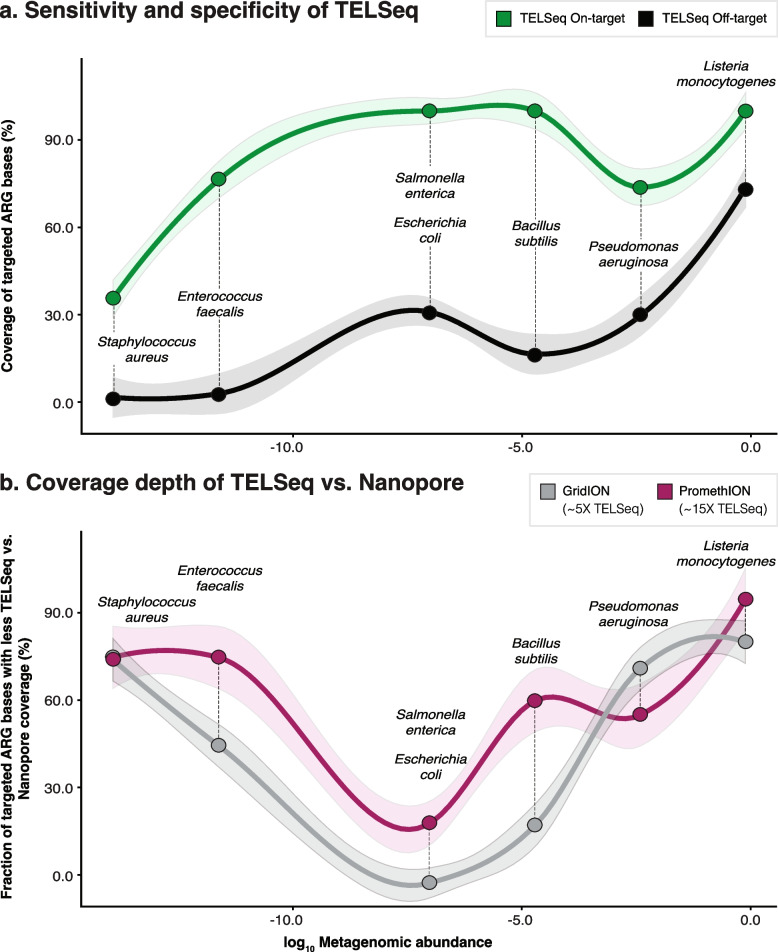
Results: Using technical replicates of diverse sample types, we compared TELSeq performance to that of non-enriched PacBio and short-read Illumina sequencing. TELSeq achieved much higher ARG recovery (>1,000-fold) and sensitivity than the other methods across diverse metagenomes, revealing an extensive resistome profile comprising many low-abundance ARGs, including some with public health importance. Using the long reads generated by TELSeq, we identified numerous MGEs and cargo genes flanking the low-abundance ARGs, indicating that these ARGs could be transferred across bacterial taxa via HGT.
Conclusions: TELSeq can provide a nuanced view of the genomic context of microbial resistomes and thus has wide-ranging applications in public, animal, and human health, as well as environmental surveillance and monitoring of AMR. Thus, this technique represents a fundamental advancement for microbiome research and application. Video abstract.
Keywords: Antimicrobial resistance; Long-read sequencing; Metagenomics; Microbiome; Mobile genetic elements; Public health; Resistome.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
