

Insights into the potential for mutualistic and harmful host-microbe interactions affecting brown alga freshwater acclimation
- PMID: 36326449
- PMCID: PMC10099861
- DOI: 10.1111/mec.16766
Insights into the potential for mutualistic and harmful host-microbe interactions affecting brown alga freshwater acclimation
Abstract
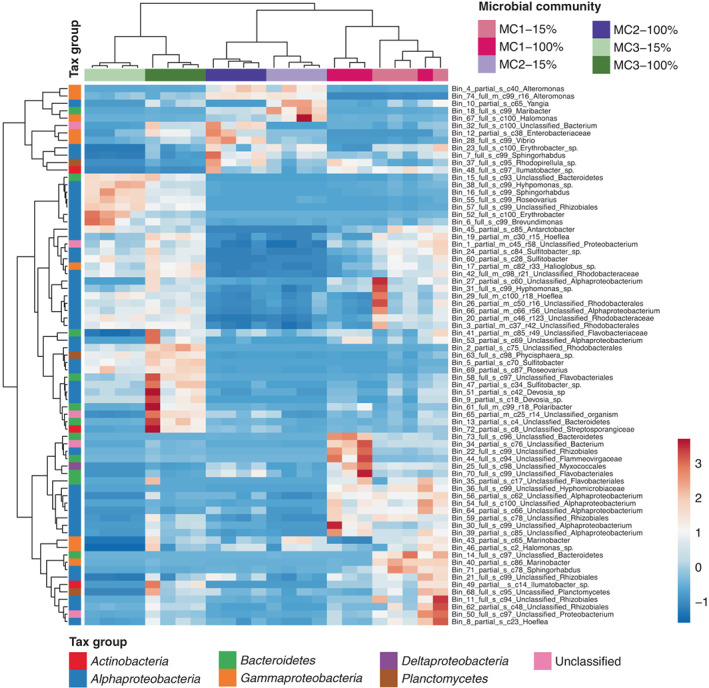
Microbes can modify their hosts' stress tolerance, thus potentially enhancing their ecological range. An example of such interactions is Ectocarpus subulatus, one of the few freshwater-tolerant brown algae. This tolerance is partially due to its (un)cultivated microbiome. We investigated this phenomenon by modifying the microbiome of laboratory-grown E. subulatus using mild antibiotic treatments, which affected its ability to grow in low salinity. Low salinity acclimation of these algal-bacterial associations was then compared. Salinity significantly impacted bacterial and viral gene expression, albeit in different ways across algal-bacterial communities. In contrast, gene expression of the host and metabolite profiles were affected almost exclusively in the freshwater-intolerant algal-bacterial communities. We found no evidence of bacterial protein production that would directly improve algal stress tolerance. However, vitamin K synthesis is one possible bacterial service missing specifically in freshwater-intolerant cultures in low salinity. In this condition, we also observed a relative increase in bacterial transcriptomic activity and the induction of microbial genes involved in the biosynthesis of the autoinducer AI-1, a quorum-sensing regulator. This could have resulted in dysbiosis by causing a shift in bacterial behaviour in the intolerant algal-bacterial community. Together, these results provide two promising hypotheses to be examined by future targeted experiments. Although they apply only to the specific study system, they offer an example of how bacteria may impact their host's stress response.
Keywords: brown algae; holobiont; host-microbiome interaction; low salinity acclimation; meta-transcriptomics; metabolic networks; metabolite profiling; virome.
© 2022 The Authors. Molecular Ecology published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures







References
-
- Aite, M. , Chevallier, M. , Frioux, C. , Trottier, C. , Got, J. , Cortés, M. P. , Mendoza, S. N. , Carrier, G. , Dameron, O. , Guillaudeux, N. , Latorre, M. , Loira, N. , Markov, G. V. , Maass, A. , & Siegel, A. (2018). Traceability, reproducibility and wiki‐exploration for “à‐la‐carte” reconstructions of genome‐scale metabolic models. PLoS Computational Biology, 14(5), e1006146. 10.1371/journal.pcbi.1006146 - DOI - PMC - PubMed
-
- Berland, B. R. , Bonin, D. J. , Guérin‐Ancey, O. , & Antia, N. J. (1979). Concentration requirement of glycine as nitrogen source for supporting effective growth of certain marine microplanktonic algae. Marine Biology, 55(2), 83–92. 10.1007/BF00397303 - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
