

Glycosylated clusterin species facilitate Aβ toxicity in human neurons
- PMID: 36329114
- PMCID: PMC9633591
- DOI: 10.1038/s41598-022-23167-z
Glycosylated clusterin species facilitate Aβ toxicity in human neurons
Abstract
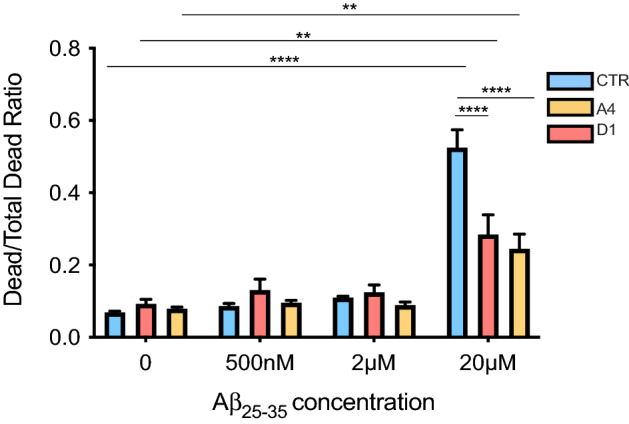
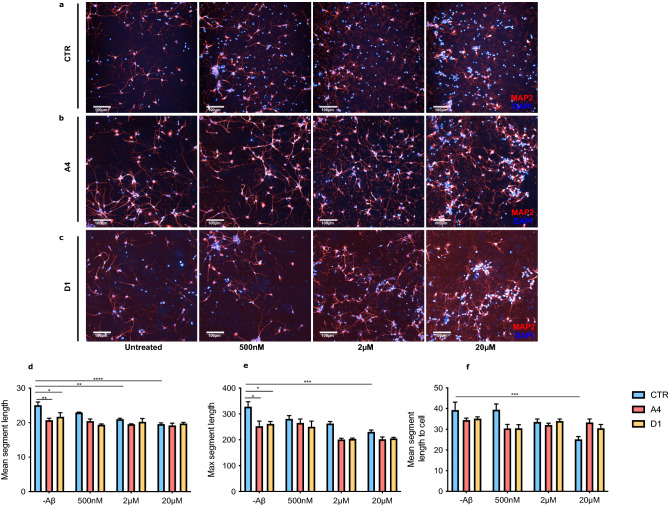
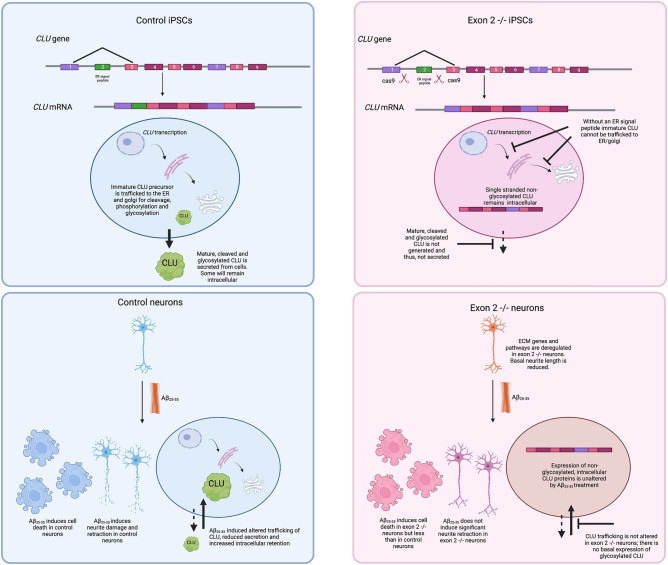
Clusterin (CLU) is one of the most significant genetic risk factors for late onset Alzheimer's disease (AD). However, the mechanisms by which CLU contributes to AD development and pathogenesis remain unclear. Studies have demonstrated that the trafficking and localisation of glycosylated CLU proteins is altered by CLU-AD mutations and amyloid-β (Aβ), which may contribute to AD pathogenesis. However, the roles of non-glycosylated and glycosylated CLU proteins in mediating Aβ toxicity have not been studied in human neurons. iPSCs with altered CLU trafficking were generated following the removal of CLU exon 2 by CRISPR/Cas9 gene editing. Neurons were generated from control (CTR) and exon 2 -/- edited iPSCs and were incubated with aggregated Aβ peptides. Aβ induced changes in cell death and neurite length were quantified to determine if altered CLU protein trafficking influenced neuronal sensitivity to Aβ. Finally, RNA-Seq analysis was performed to identify key transcriptomic differences between CLU exon 2 -/- and CTR neurons. The removal of CLU exon 2, and the endoplasmic reticulum (ER)-signal peptide located within, abolished the presence of glycosylated CLU and increased the abundance of intracellular, non-glycosylated CLU. While non-glycosylated CLU levels were unaltered by Aβ25-35 treatment, the trafficking of glycosylated CLU was altered in control but not exon 2 -/- neurons. The latter also displayed partial protection against Aβ-induced cell death and neurite retraction. Transcriptome analysis identified downregulation of multiple extracellular matrix (ECM) related genes in exon 2 -/- neurons, potentially contributing to their reduced sensitivity to Aβ toxicity. This study identifies a crucial role of glycosylated CLU in facilitating Aβ toxicity in human neurons. The loss of these proteins reduced both, cell death and neurite damage, two key consequences of Aβ toxicity identified in the AD brain. Strikingly, transcriptomic differences between exon 2 -/- and control neurons were small, but a significant and consistent downregulation of ECM genes and pathways was identified in exon 2 -/- neurons. This may contribute to the reduced sensitivity of these neurons to Aβ, providing new mechanistic insights into Aβ pathologies and therapeutic targets for AD.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
Clusterin Is Required for β-Amyloid Toxicity in Human iPSC-Derived Neurons.Front Neurosci. 2018 Jul 25;12:504. doi: 10.3389/fnins.2018.00504. eCollection 2018. Front Neurosci. 2018. PMID: 30090055 Free PMC article.
-
Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer's disease neuropathology.Mol Neurodegener. 2021 Jan 31;16(1):5. doi: 10.1186/s13024-021-00426-7. Mol Neurodegener. 2021. PMID: 33517893 Free PMC article.
-
Intracellular and secreted forms of clusterin are elevated early in Alzheimer's disease and associate with both Aβ and tau pathology.Neurobiol Aging. 2020 May;89:129-131. doi: 10.1016/j.neurobiolaging.2019.10.025. Epub 2019 Nov 8. Neurobiol Aging. 2020. PMID: 31813628
-
Exploring the Role of CLU in the Pathogenesis of Alzheimer's Disease.Neurotox Res. 2021 Dec;39(6):2108-2119. doi: 10.1007/s12640-020-00271-4. Epub 2020 Aug 21. Neurotox Res. 2021. Retraction in: Neurotox Res. 2022 Aug;40(4):1125. doi: 10.1007/s12640-022-00519-1. PMID: 32820456 Retracted. Review.
-
Clusterin in Alzheimer's Disease: Mechanisms, Genetics, and Lessons From Other Pathologies.Front Neurosci. 2019 Feb 28;13:164. doi: 10.3389/fnins.2019.00164. eCollection 2019. Front Neurosci. 2019. PMID: 30872998 Free PMC article. Review.
Cited by
-
Changes in iPSC-astrocyte morphology reflect Alzheimer's disease patient clinical markers.Stem Cells. 2025 Mar 10;43(3):sxae085. doi: 10.1093/stmcls/sxae085. Stem Cells. 2025. PMID: 39704342 Free PMC article.
-
The decreased astrocyte-microglia interaction reflects the early characteristics of Alzheimer's disease.iScience. 2024 Feb 20;27(3):109281. doi: 10.1016/j.isci.2024.109281. eCollection 2024 Mar 15. iScience. 2024. PMID: 38455972 Free PMC article.
-
Glycoproteomics in Cerebrospinal Fluid Reveals Brain-Specific Glycosylation Changes.Int J Mol Sci. 2023 Jan 18;24(3):1937. doi: 10.3390/ijms24031937. Int J Mol Sci. 2023. PMID: 36768261 Free PMC article.
-
Clusterin/apolipoprotein J, its isoforms and Alzheimer's disease.Front Aging Neurosci. 2023 Apr 13;15:1167886. doi: 10.3389/fnagi.2023.1167886. eCollection 2023. Front Aging Neurosci. 2023. PMID: 37122381 Free PMC article. No abstract available.
-
Brain and plasmatic CLUSTERIN are translational markers of Alzheimer's disease.Brain Pathol. 2025 Mar;35(2):e13281. doi: 10.1111/bpa.13281. Epub 2024 Jun 22. Brain Pathol. 2025. PMID: 39965636 Free PMC article.
References
-
- National Institute on Aging. Alzheimer’s Disease Fact Sheet. http://www.nia.nih.gov/health/alzheimers-disease-fact-sheet (2019).
-
- Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. - PubMed
-
- Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
