

Advances and limitations for the treatment of spinal muscular atrophy
- PMID: 36329412
- PMCID: PMC9632131
- DOI: 10.1186/s12887-022-03671-x
Advances and limitations for the treatment of spinal muscular atrophy
Abstract
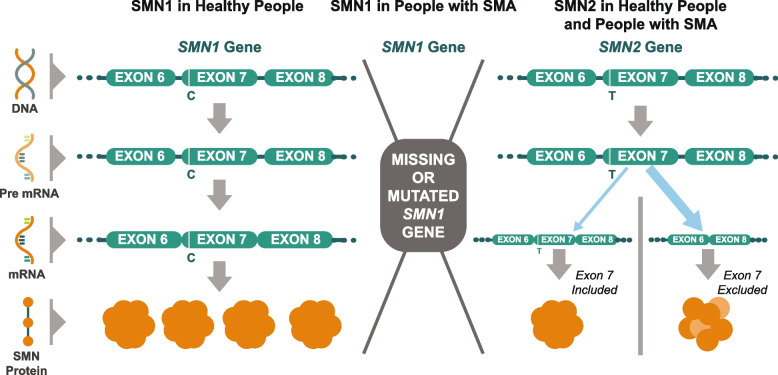
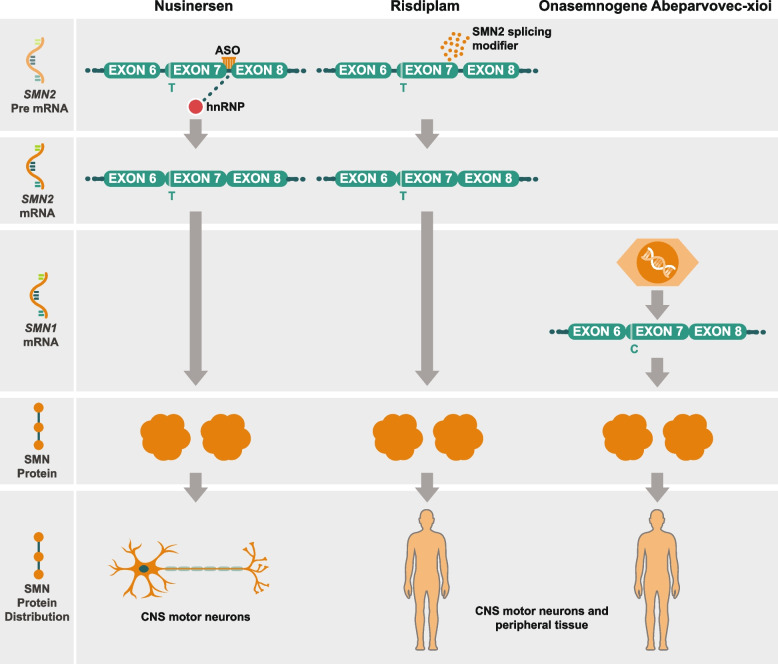
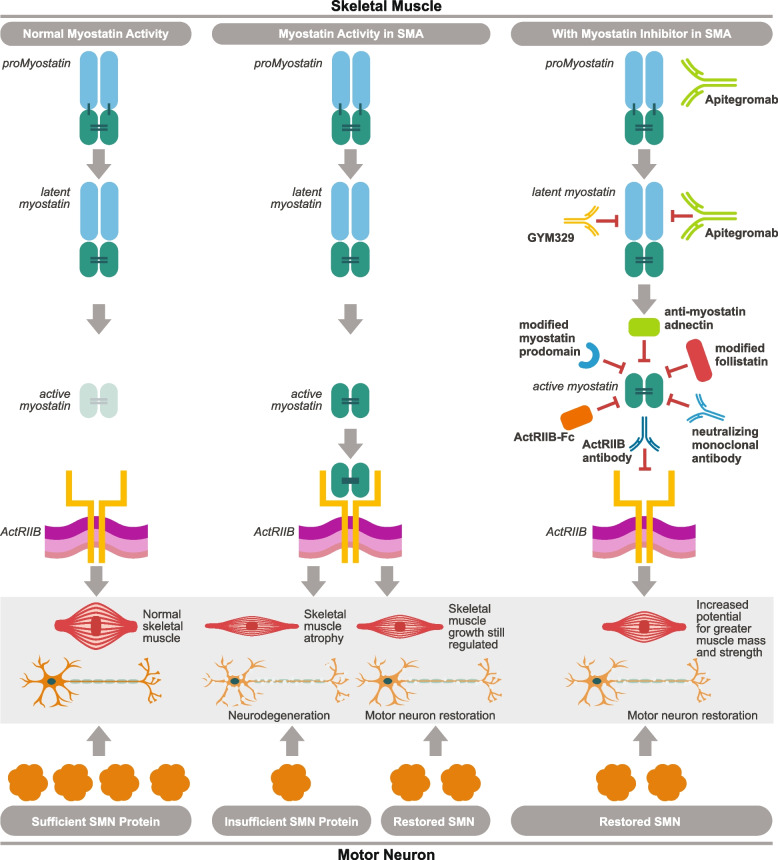
Spinal muscular atrophy (5q-SMA; SMA), a genetic neuromuscular condition affecting spinal motor neurons, is caused by defects in both copies of the SMN1 gene that produces survival motor neuron (SMN) protein. The highly homologous SMN2 gene primarily expresses a rapidly degraded isoform of SMN protein that causes anterior horn cell degeneration, progressive motor neuron loss, skeletal muscle atrophy and weakness. Severe cases result in limited mobility and ventilatory insufficiency. Untreated SMA is the leading genetic cause of death in young children. Recently, three therapeutics that increase SMN protein levels in patients with SMA have provided incremental improvements in motor function and developmental milestones and prevented the worsening of SMA symptoms. While the therapeutic approaches with Spinraza®, Zolgensma®, and Evrysdi® have a clinically significant impact, they are not curative. For many patients, there remains a significant disease burden. A potential combination therapy under development for SMA targets myostatin, a negative regulator of muscle mass and strength. Myostatin inhibition in animal models increases muscle mass and function. Apitegromab is an investigational, fully human, monoclonal antibody that specifically binds to proforms of myostatin, promyostatin and latent myostatin, thereby inhibiting myostatin activation. A recently completed phase 2 trial demonstrated the potential clinical benefit of apitegromab by improving or stabilizing motor function in patients with Type 2 and Type 3 SMA and providing positive proof-of-concept for myostatin inhibition as a target for managing SMA. The primary goal of this manuscript is to orient physicians to the evolving landscape of SMA treatment.
Keywords: Apitegromab; Myostatin; Nusinersen; Onasemnogene abeparvovec-xioi; Risdiplam; SRK-015; Spinal muscular atrophy; Survival motor neuron; Survival motor neuron-1 gene.
© 2022. The Author(s).
Conflict of interest statement
Drs. George Nomikos, Nathalie Kertesz and Jose Rossello are employees of Scholar Rock, Inc. and are shareholders. Drs. Amy Place and Kimberly Long are former employees of Scholar Rock. Dr. John Day reports grants from: AMO Pharma; Audentes; Avidity; Biogen; Cytokinetics; Genentech; Ionis Pharmaceuticals; Novartis Gene Therapies; Roche Pharmaceuticals; Sanofi-Genzyme; Sarepta Therapeutics; Scholar Rock, and has received support from participating on advisory boards or having consulted with: Affinia Therapeutics; AMO Pharmaceuticals; Avidity Biosciences; Biogen; Cytokinetics; Epirium Bio; Ionis Pharmaceuticals; Kate Therapeutics; Novartis Gene Therapies; Roche/Genentech Pharmaceuticals; Sarepta Therapeutics; Scholar Rock; Shift Therapeutics; and Vertex Pharmaceuticals. Dr Kelly Howell has nothing to disclose.
Figures
References
-
- Cure SMA. Elk Grove Village, IL. https://www.curesma.org/about-sma/. Accessed 18 July 2021.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
