

Microtubule retrograde flow retains neuronal polarization in a fluctuating state
- PMID: 36332023
- PMCID: PMC9635824
- DOI: 10.1126/sciadv.abo2336
Microtubule retrograde flow retains neuronal polarization in a fluctuating state
Abstract
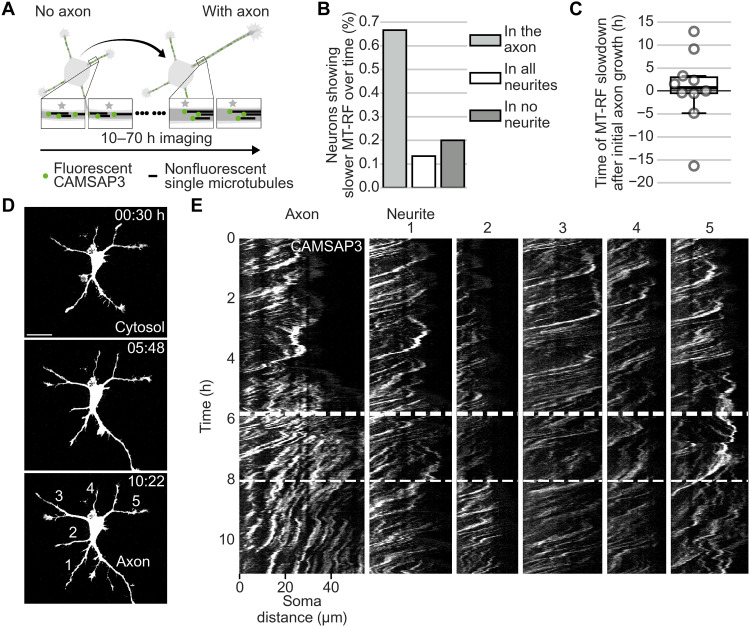
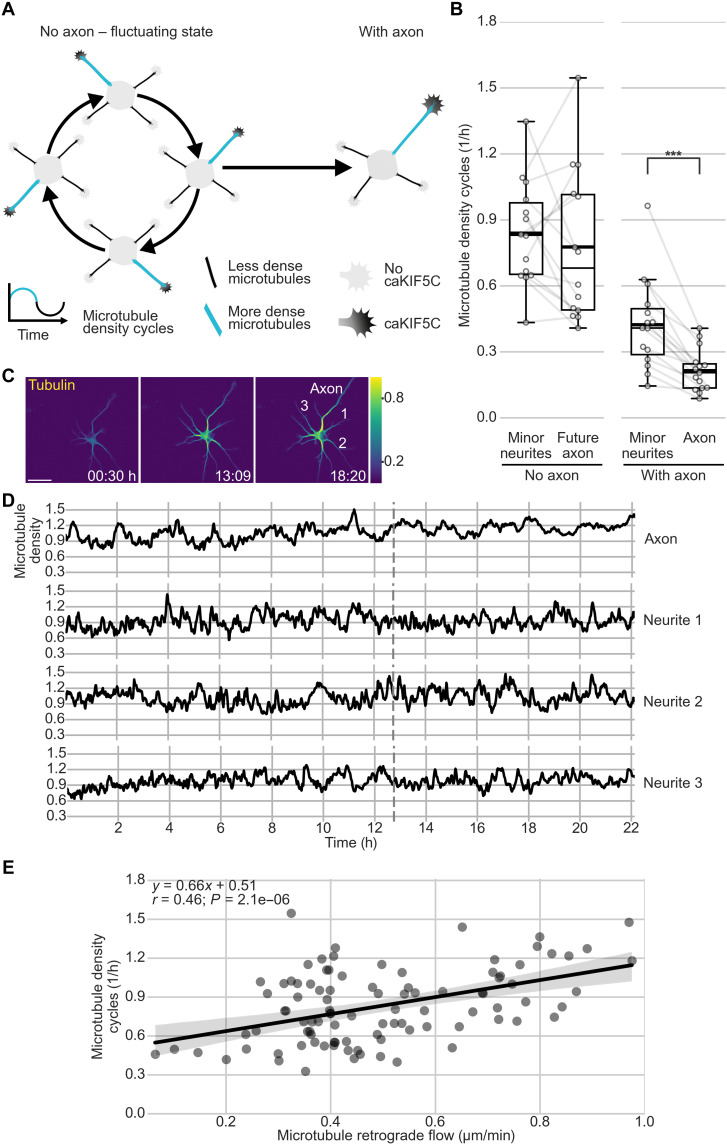
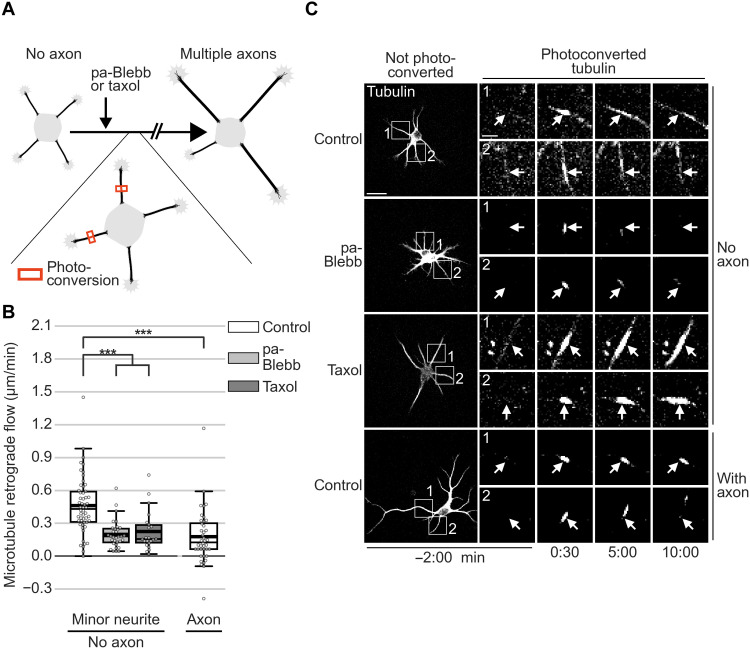
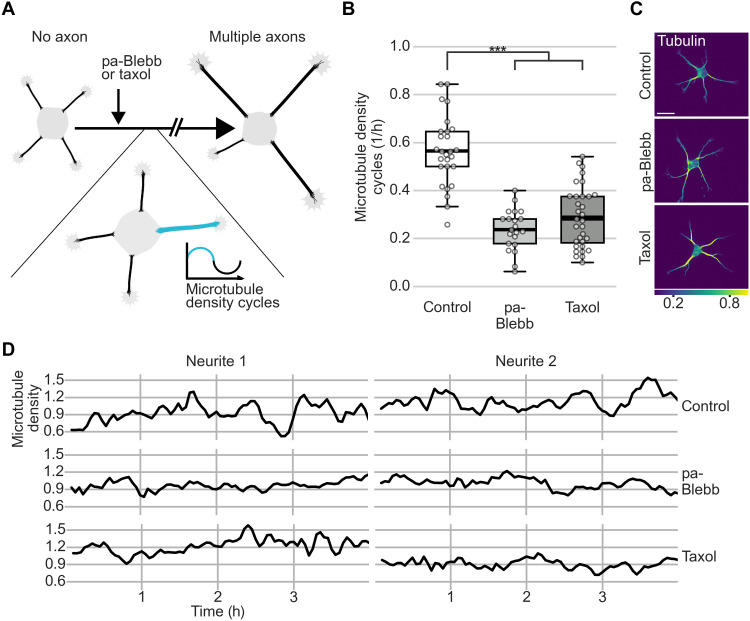
In developing vertebrate neurons, a neurite is formed by more than a hundred microtubules. While individual microtubules are dynamic, the microtubule array has been regarded as stationary. Using live-cell imaging of neurons in culture or in brain slices, combined with photoconversion techniques and pharmacological manipulations, we uncovered that the microtubule array flows retrogradely within neurites to the soma. This flow drives cycles of microtubule density, a hallmark of the fluctuating state before axon formation, thereby inhibiting neurite growth. The motor protein dynein fuels this process. Shortly after axon formation, microtubule retrograde flow slows down in the axon, reducing microtubule density cycles and enabling axon extension. Thus, keeping neurites short is an active process. Microtubule retrograde flow is a previously unknown type of cytoskeletal dynamics, which changes the hitherto axon-centric view of neuronal polarization.
Figures






Similar articles
-
Local changes in microtubule network mobility instruct neuronal polarization and axon specification.Sci Adv. 2022 Nov 4;8(44):eabo2343. doi: 10.1126/sciadv.abo2343. Epub 2022 Nov 4. Sci Adv. 2022. PMID: 36332030 Free PMC article.
-
Dendrites In Vitro and In Vivo Contain Microtubules of Opposite Polarity and Axon Formation Correlates with Uniform Plus-End-Out Microtubule Orientation.J Neurosci. 2016 Jan 27;36(4):1071-85. doi: 10.1523/JNEUROSCI.2430-15.2016. J Neurosci. 2016. PMID: 26818498 Free PMC article.
-
Laminin/β1 integrin signal triggers axon formation by promoting microtubule assembly and stabilization.Cell Res. 2012 Jun;22(6):954-72. doi: 10.1038/cr.2012.40. Epub 2012 Mar 20. Cell Res. 2012. PMID: 22430151 Free PMC article.
-
Spastin Interacts with CRMP2 to Regulate Neurite Outgrowth by Controlling Microtubule Dynamics through Phosphorylation Modifications.CNS Neurol Disord Drug Targets. 2021 Oct 26;20(3):249-265. doi: 10.2174/1871527319666201026165855. CNS Neurol Disord Drug Targets. 2021. PMID: 33109053 Review.
-
An Integrated Cytoskeletal Model of Neurite Outgrowth.Front Cell Neurosci. 2018 Nov 26;12:447. doi: 10.3389/fncel.2018.00447. eCollection 2018. Front Cell Neurosci. 2018. PMID: 30534055 Free PMC article. Review.
Cited by
-
Nesprin-2 coordinates opposing microtubule motors during nuclear migration in neurons.J Cell Biol. 2024 Nov 4;223(11):e202405032. doi: 10.1083/jcb.202405032. Epub 2024 Aug 8. J Cell Biol. 2024. PMID: 39115447 Free PMC article.
-
Axonal Mechanotransduction Drives Cytoskeletal Responses to Physiological Mechanical Forces.bioRxiv [Preprint]. 2025 Feb 12:2025.02.11.637689. doi: 10.1101/2025.02.11.637689. bioRxiv. 2025. PMID: 39990487 Free PMC article. Preprint.
-
Age-dependent increase of cytoskeletal components in sensory axons in human skin.Front Cell Dev Biol. 2022 Nov 1;10:965382. doi: 10.3389/fcell.2022.965382. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36393849 Free PMC article.
-
Mechanisms of Axon Growth and Regeneration: Moving between Development and Disease.J Neurosci. 2022 Nov 9;42(45):8393-8405. doi: 10.1523/JNEUROSCI.1131-22.2022. J Neurosci. 2022. PMID: 36351827 Free PMC article.
-
Cellular cartography: Towards an atlas of the neuronal microtubule cytoskeleton.Front Cell Dev Biol. 2023 Mar 22;11:1052245. doi: 10.3389/fcell.2023.1052245. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37035244 Free PMC article. Review.
References
-
- Kapitein L. C., Hoogenraad C. C., Building the neuronal microtubule cytoskeleton. Neuron 87, 492–506 (2015). - PubMed
-
- Baas P. W., Nadar C. V., Myers K. A., Axonal transport of microtubules: The long and short of It. Traffic 7, 490–498 (2006). - PubMed
-
- Lüders J., Nucleating microtubules in neurons: Challenges and solutions. Dev. Neurobiol. 81, 273–283 (2020). - PubMed
-
- Stiess M., Maghelli N., Kapitein L. C., Gomis-Rüth S., Wilsch-Bräuninger M., Hoogenraad C. C., Tolić-Nørrelykke I. M., Bradke F., Axon extension occurs independently of centrosomal microtubule nucleation. Science 327, 704–707 (2010). - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
