

MOSAICS: A software suite for analysis of membrane structure and dynamics in simulated trajectories
- PMID: 36333911
- PMCID: PMC10257019
- DOI: 10.1016/j.bpj.2022.11.005
MOSAICS: A software suite for analysis of membrane structure and dynamics in simulated trajectories
Abstract
Molecular dynamics (MD) simulations have become the predominant computational analysis method in membrane biophysics, as this technique is uniquely suited for investigations of complex molecular systems through the relevant physical principles. Owing to continued improvements in scope and performance, the trajectories generated through this approach contain ever-increasing amounts of information, which must be synthesized and simplified in post-analysis using tools that are not only mechanistically insightful but also computationally efficient and highly scalable. Here, we introduce MOSAICS, a self-contained high-performance suite of C++ software tools designed for advanced analyses of lipid bilayer structure and dynamics from MD trajectories. MOSAICS is to our knowledge the most comprehensive software suite of this kind, enabling analysis of a wide array of morphological and kinetic properties, for both simple and complex membranes, irrespective of system size or resolution. Importantly, MOSAICS is designed to provide spatial distributions of all computed quantities, with built-in masking tools, noise filtering, and statistical significance metrics to facilitate quantitative interpretations of the trajectory data; it is also fully parallelized and can therefore leverage the capabilities of supercomputing facilities. Despite its technical sophistication, MOSAICS is user-friendly and requires minimal computational expertise, making it accessible to researchers of all skill levels. This sofware suite can be freely downloaded at https://github.com/MOSAICS-NIH/.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
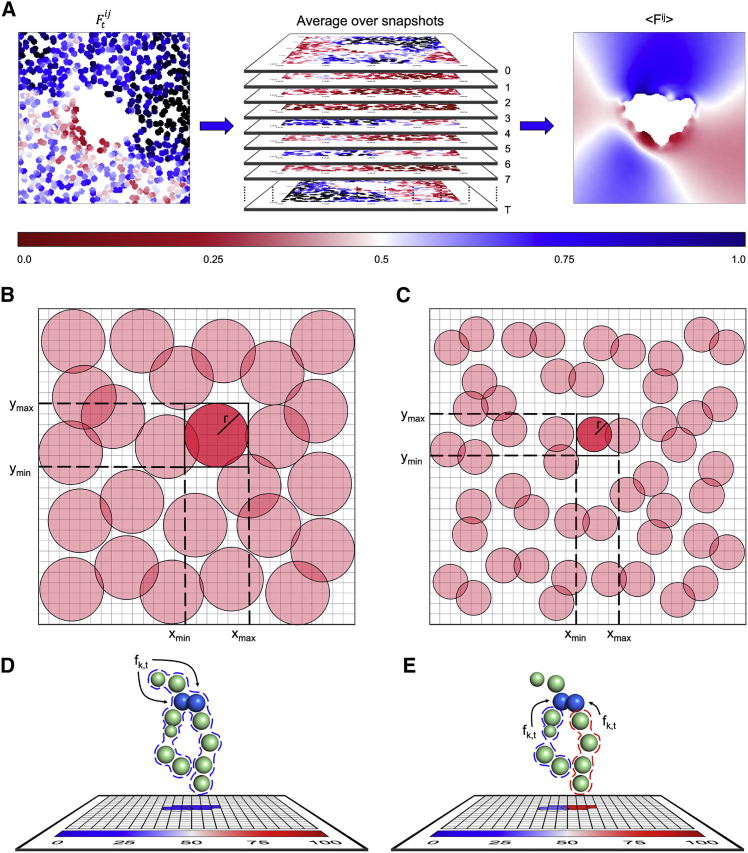

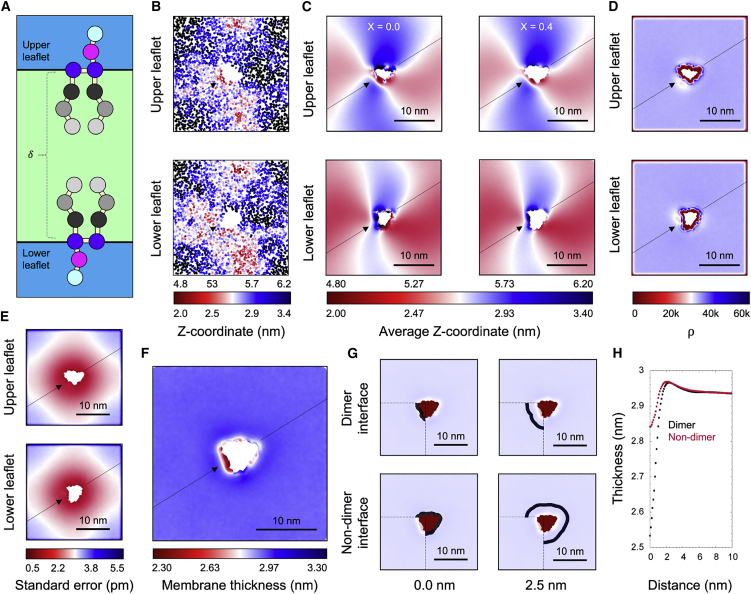

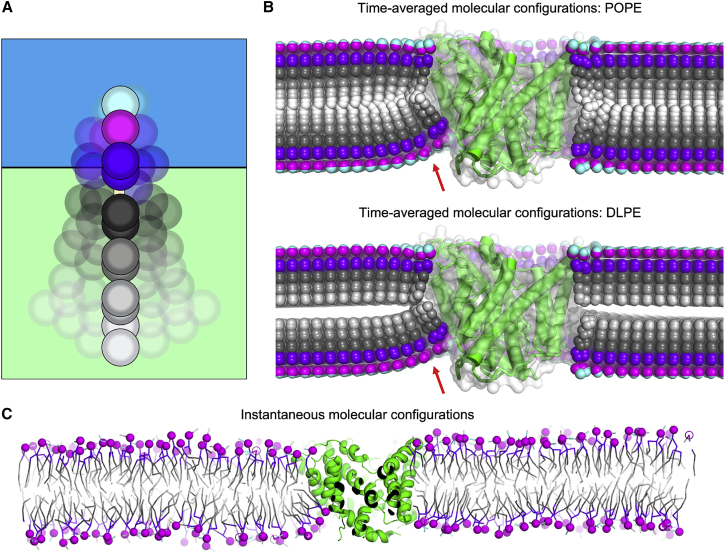




References
-
- Allen W.J., Lemkul J.A., Bevan D.R. GridMAT-MD: a grid-based membrane analysis tool for use with molecular dynamics. J. Comput. Chem. 2009;30:1952–1958. - PubMed
-
- Lukat G., Krüger J., Sommer B. APL@Voro: a Voronoi-based membrane analysis tool for GROMACS trajectories. J. Chem. Inf. Model. 2013;53:2908–2925. - PubMed
-
- Ramasubramani V., Dice B.D., et al. Glotzer S.C. Freud: a software suite for high throughput analysis of particle simulation data. Comput. Phys. Commun. 2020;254:107275.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
