

The Dynamics of Adaptation to Stress from Standing Genetic Variation and de novo Mutations
- PMID: 36334099
- PMCID: PMC9703598
- DOI: 10.1093/molbev/msac242
The Dynamics of Adaptation to Stress from Standing Genetic Variation and de novo Mutations
Abstract
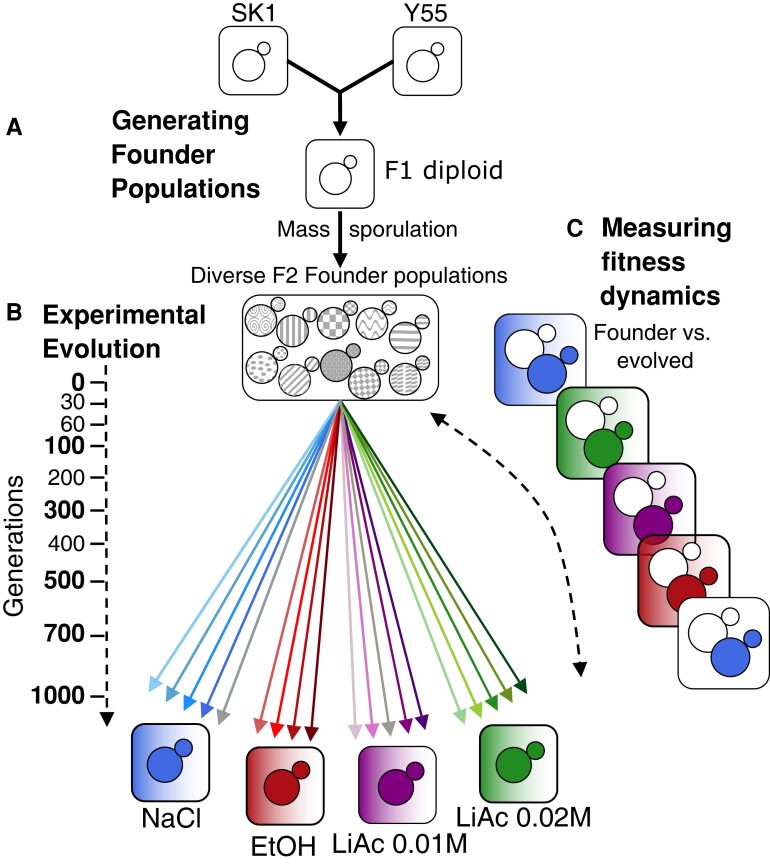
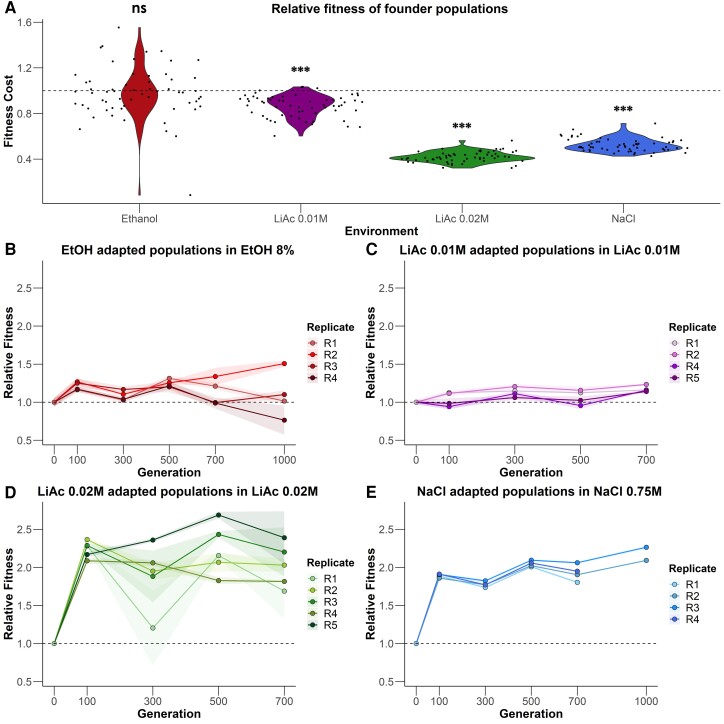
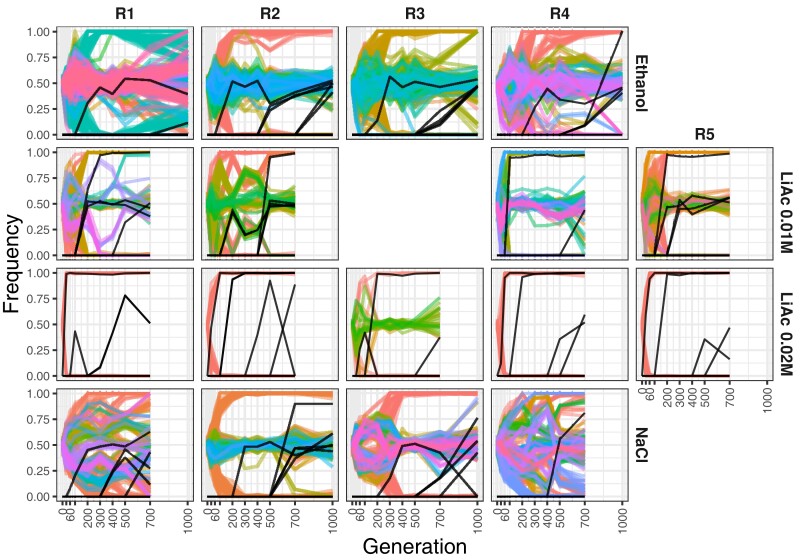
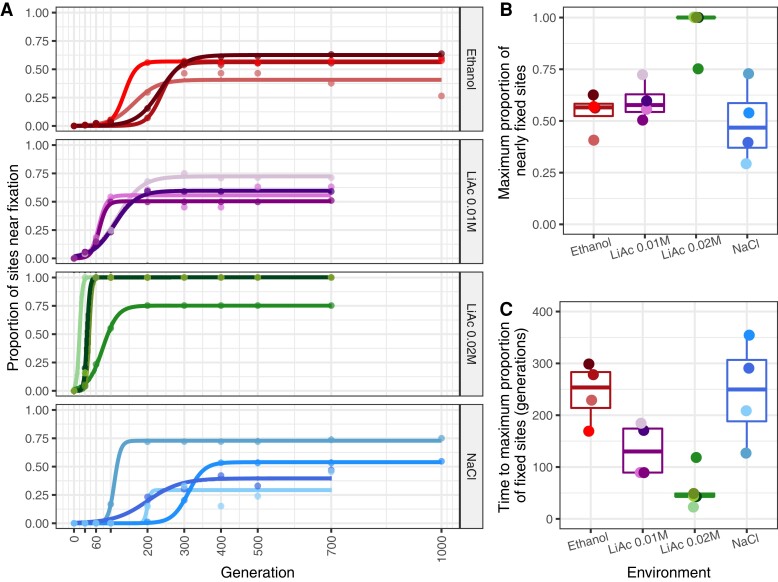
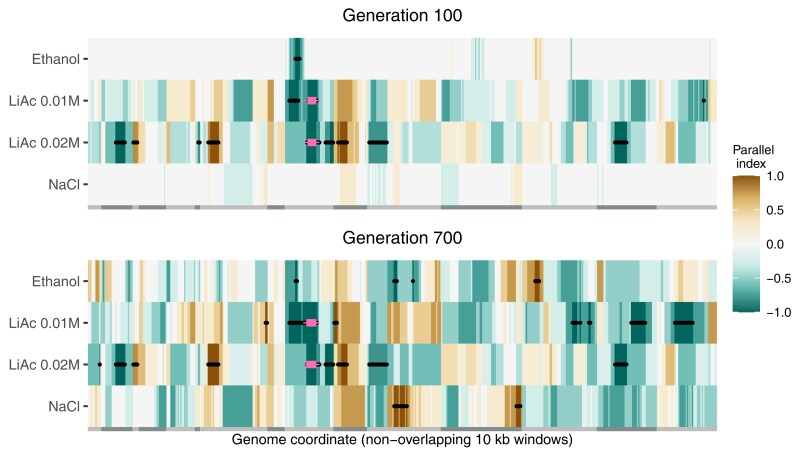
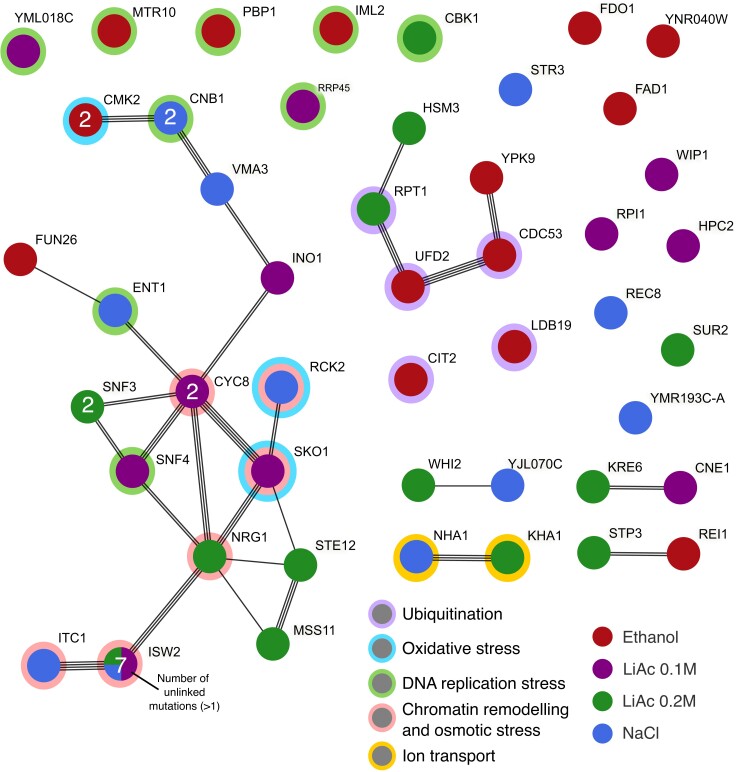
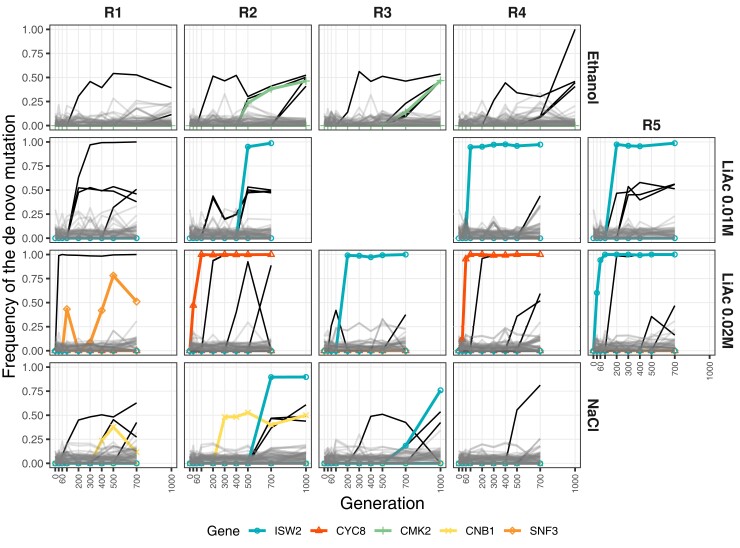
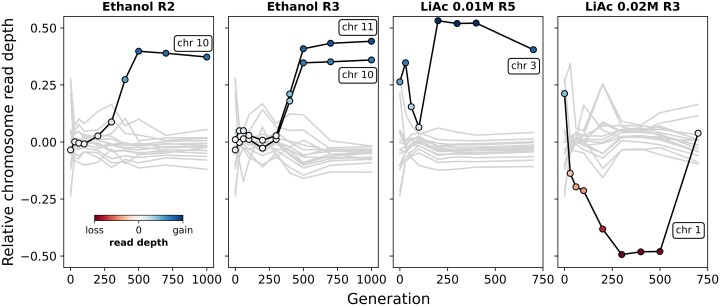
Adaptation from standing genetic variation is an important process underlying evolution in natural populations, but we rarely get the opportunity to observe the dynamics of fitness and genomic changes in real time. Here, we used experimental evolution and Pool-Seq to track the phenotypic and genomic changes of genetically diverse asexual populations of the yeast Saccharomyces cerevisiae in four environments with different fitness costs. We found that populations rapidly and in parallel increased in fitness in stressful environments. In contrast, allele frequencies showed a range of trajectories, with some populations fixing all their ancestral variation in <30 generations and others maintaining diversity across hundreds of generations. We detected parallelism at the genomic level (involving genes, pathways, and aneuploidies) within and between environments, with idiosyncratic changes recurring in the environments with higher stress. In particular, we observed a tendency of becoming haploid-like in one environment, whereas the populations of another environment showed low overall parallelism driven by standing genetic variation despite high selective pressure. This work highlights the interplay between standing genetic variation and the influx of de novo mutations in populations adapting to a range of selective pressures with different underlying trait architectures, advancing our understanding of the constraints and drivers of adaptation.
Keywords: Pool-Seq; adaptation dynamics; environmental stress; microbial experimental evolution; parallelism; time series; yeast.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
-
- Andrews S. 2010. FASTQC. A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
-
- Auguie B. 2017. gridExtra: Miscellaneous Functions for “Grid” Graphics.
-
- Bailey SF, Blanquart F, Bataillon T, Kassen R. 2017. What drives parallel evolution?: how population size and mutational variation contribute to repeated evolution. BioEssays 39(1):e201600176. - PubMed
-
- Bailey SF, Rodrigue N, Kassen R. 2015. The effect of selection environment on the probability of parallel evolution. Mol Biol Evol. 32(6):1436–1448. - PubMed
-
- Barghi N, Hermisson J, Schlötterer C. 2020. Polygenic adaptation: a unifying framework to understand positive selection. Nat Rev Genet. 21(12):769–781. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
