

Nanoparticle endothelial delivery of PGC-1α attenuates hypoxia-induced pulmonary hypertension by attenuating EndoMT-caused vascular wall remodeling
- PMID: 36334380
- PMCID: PMC9637961
- DOI: 10.1016/j.redox.2022.102524
Nanoparticle endothelial delivery of PGC-1α attenuates hypoxia-induced pulmonary hypertension by attenuating EndoMT-caused vascular wall remodeling
Abstract
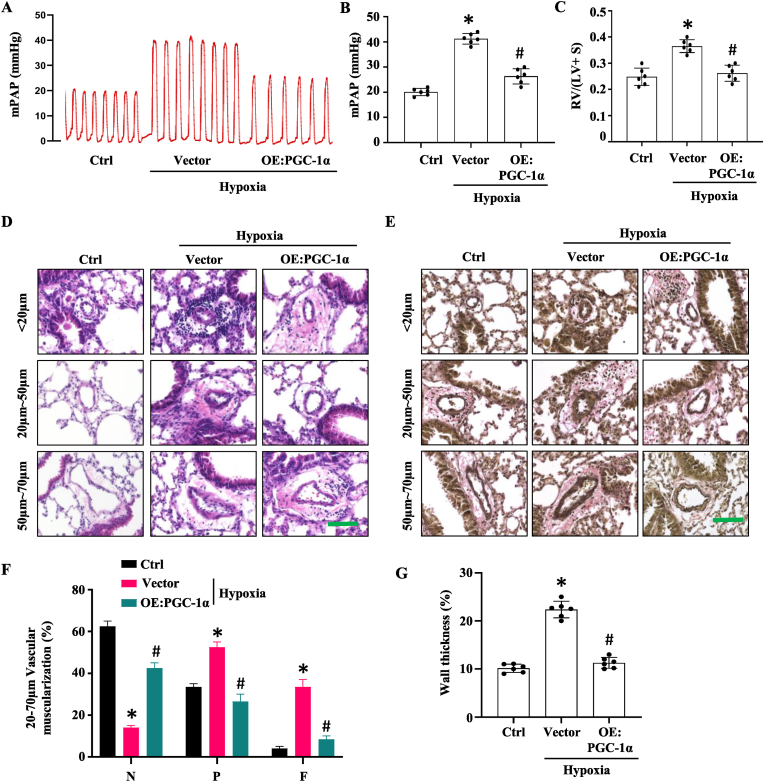
Pulmonary hypertension (PH) induced by chronic hypoxia is characterized by thickening of pulmonary artery walls, elevated pulmonary vascular resistance, and right-heart failure. Dysfunction of endothelial cells is the hallmark event in the progression of PH. Among various mechanisms, endothelial to mesenchymal transition (EndoMT) has emerged as an important source of endothelial cell dysfunction in PH. However, the mechanisms underlying the EndoMT in PH remain largely unknown. Our results showed that peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) expression was decreased in pulmonary arterial endothelial cells (PAECs) in PH patients and hypoxia-induced PH mouse model compared to the normal controls. Endothelial-specific overexpression of PGC-1α using nanoparticle delivery significantly attenuated the progression of PH, as shown by the significantly decreased right ventricular systolic pressure and diminished artery thickness as well as reduced vascular muscularization. Moreover, Endothelial-specific overexpression of PGC-1α blocked the EndoMT of PAECs during PH, indicating that loss of PGC-1α promotes PH development by mediating EndoMT, which damages the integrity of endothelium. Intriguingly, we found that PGC-1α overexpression rescued the expression of endothelial nitric oxide synthase in mouse lung tissues that was deceased by hypoxia treatment in vivo and in endothelial cells treated with TGF-β in vitro. Consistently, PAECs and vascular smooth muscle co-culture showed that overexpression of PGC-1α in PAECs increases nitric oxide release, which would likely diffuse to smooth muscle cells, where it activates specific protein kinases, and initiates SMC relaxation by diminishing the calcium flux. Endothelial-specific overexpression of PGC-1α also attenuated hypoxia-induced pulmonary artery stiffness which appeared to be caused by both the decreased endothelial nitric oxide production and increased vascular remodeling. Taken together, these results demonstrated that endothelial-specific delivery of PGC-1α prevents PH development by inhibiting EndoMT of PAECs and thus restoring endothelial function and reducing vascular remodeling.
Keywords: Endothelial to mesenchymal transition; Nitric oxide; Peroxisome proliferator-activated receptor gamma coactivator-1α; Pulmonary hypertension.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest None.
Figures








References
-
- Chang K.Y., Duval S., Badesch D.B., Bull T.M., Chakinala M.M., De Marco T., Frantz R.P., Hemnes A., Mathai S.C., Rosenzweig E.B., Ryan J.J., Thenappan T., * P.I. Mortality in pulmonary arterial hypertension in the modern era: early insights from the pulmonary hypertension association registry. J. Am. Heart Assoc. 2022;11 - PMC - PubMed
-
- Farber H.W., Loscalzo J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004;351:1655–1665. - PubMed
-
- Tuder R.M., Stacher E., Robinson J., Kumar R., Graham B.B. Pathology of pulmonary hypertension. Clin. Chest Med. 2013;34:639–650. - PubMed
-
- Dorfmuller P. Pulmonary hypertension: pathology. Handb. Exp. Pharmacol. 2013;218:59–75. - PubMed
-
- Tuchscherer H.A., Webster E.B., Chesler N.C. Pulmonary vascular resistance and impedance in isolated mouse lungs: effects of pulmonary emboli. Ann. Biomed. Eng. 2006;34:660–668. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
