

Natural history and management of liver dysfunction in lysosomal storage disorders
- PMID: 36340750
- PMCID: PMC9627439
- DOI: 10.4254/wjh.v14.i10.1844
Natural history and management of liver dysfunction in lysosomal storage disorders
Abstract
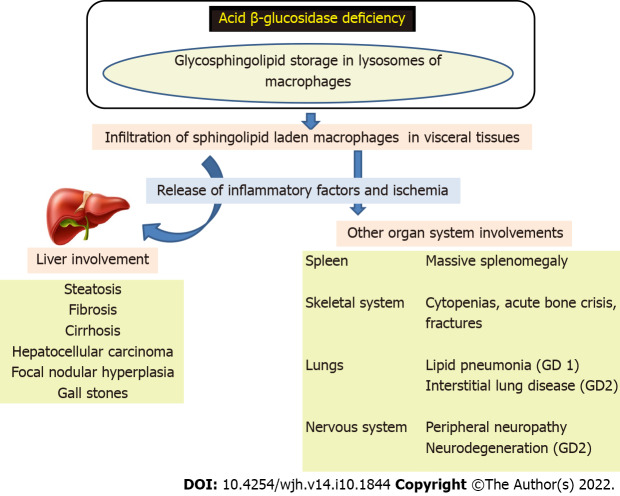
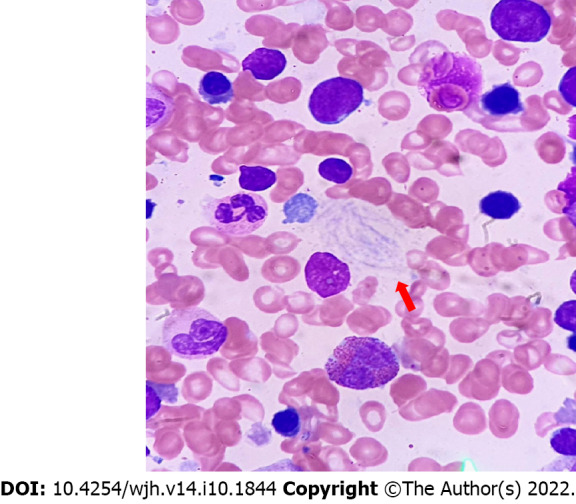
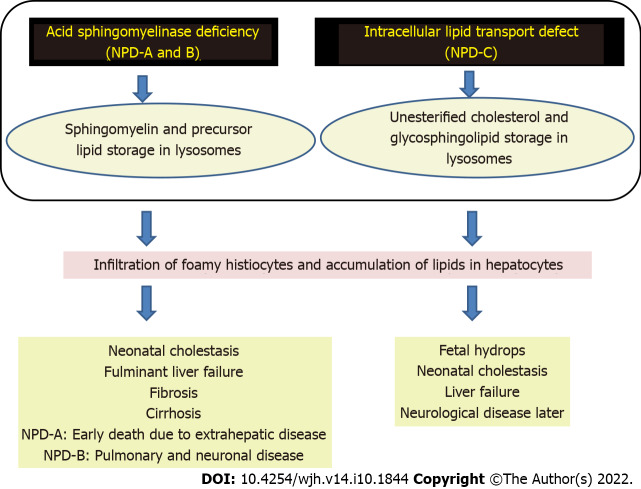
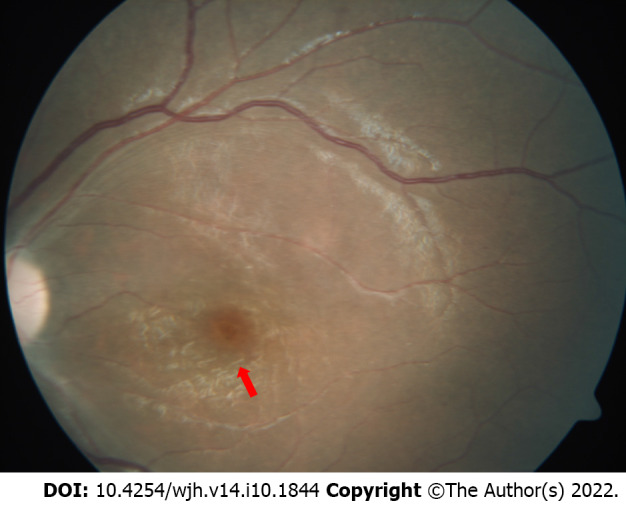
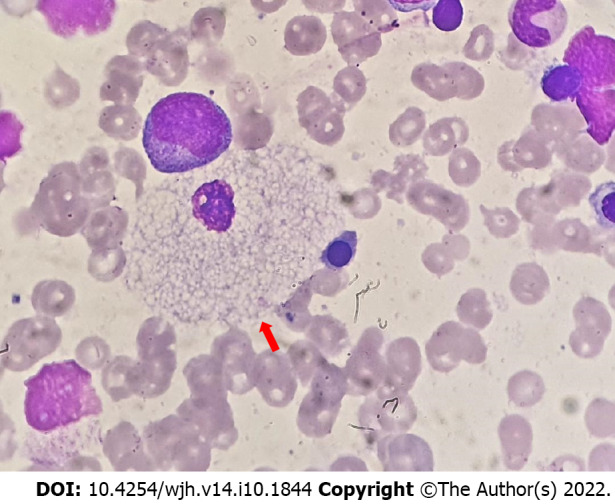
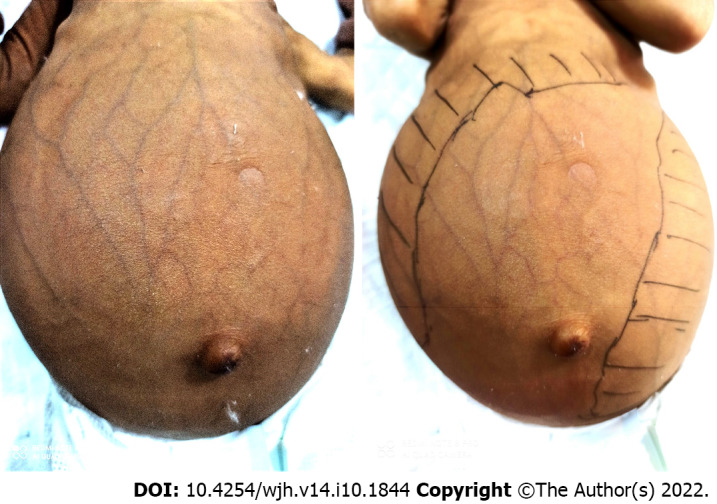
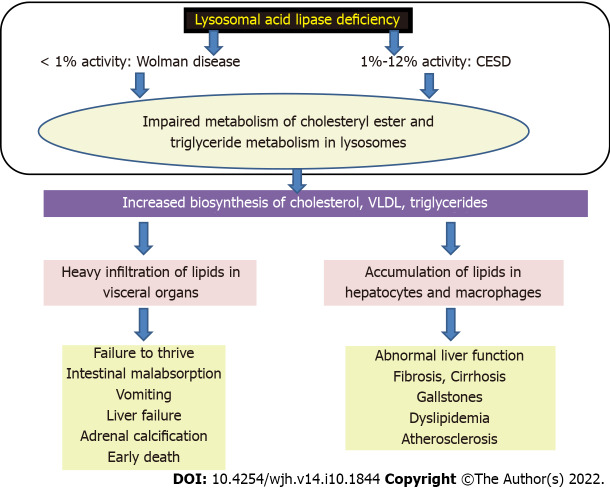
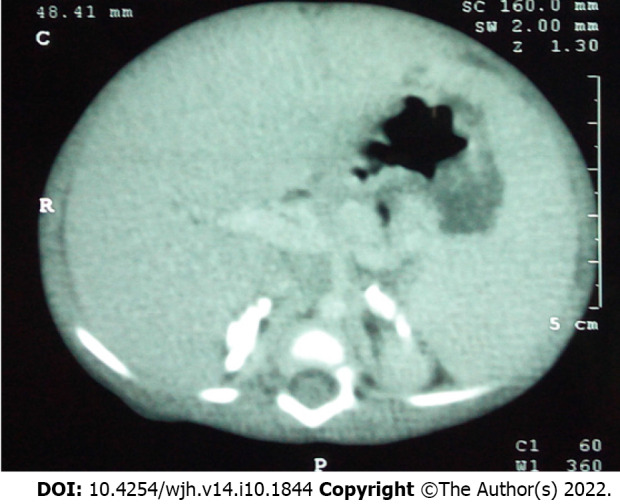
Lysosomal storage disorders (LSD) are a rare group of genetic disorders. The major LSDs that cause liver dysfunction are disorders of sphingolipid lipid storage [Gaucher disease (GD) and Niemann-Pick disease] and lysosomal acid lipase deficiency [cholesteryl ester storage disease and Wolman disease (WD)]. These diseases can cause significant liver problems ranging from asymptomatic hepatomegaly to cirrhosis and portal hypertension. Abnormal storage cells initiate hepatic fibrosis in sphingolipid disorders. Dyslipidemia causes micronodular cirrhosis in lipid storage disorders. These disorders must be keenly differentiated from other chronic liver diseases and non-alcoholic steatohepatitis that affect children and young adults. GD, Niemann-Pick type C, and WD also cause neonatal cholestasis and infantile liver failure. Genotype and liver phenotype correlation is variable in these conditions. Patients with LSD may survive up to 4-5 decades except for those with neonatal onset disease. The diagnosis of all LSD is based on enzymatic activity, tissue histology, and genetic testing. Enzyme replacement is possible in GD and Niemann-Pick types A and B though there are major limitations in the outcome. Those that progress invariably require liver transplantation with variable outcomes. The prognosis of Niemann-Pick type C and WD is universally poor. Enzyme replacement therapy has a promising role in cholesteryl ester storage disease. This review attempts to outline the natural history of these disorders from a hepatologist's perspective to increase awareness and facilitate better management of these rare disorders.
Keywords: Children; Cholesteryl ester; Gaucher; Lysosomal; Niemann-Pick; Wolman.
©The Author(s) 2022. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: No conflict of interests.
Figures
References
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105:151–156. - PubMed
-
- Cox TM, Schofield JP. Gaucher's disease: clinical features and natural history. Baillieres Clin Haematol. 1997;10:657–689. - PubMed
-
- Puri RD, Kapoor S, Kishnani PS, Dalal A, Gupta N, Muranjan M, Phadke SR, Sachdeva A, Verma IC, Mistry PK Gaucher Disease Task Force. Diagnosis and Management of Gaucher Disease in India - Consensus Guidelines of the Gaucher Disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics. Indian Pediatr. 2018;55:143–153. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
