

β-Galactosidase deficiency in the GLB1 spectrum of lysosomal storage disease can present with severe muscle weakness and atrophy
- PMID: 36341176
- PMCID: PMC9626661
- DOI: 10.1002/jmd2.12324
β-Galactosidase deficiency in the GLB1 spectrum of lysosomal storage disease can present with severe muscle weakness and atrophy
Abstract
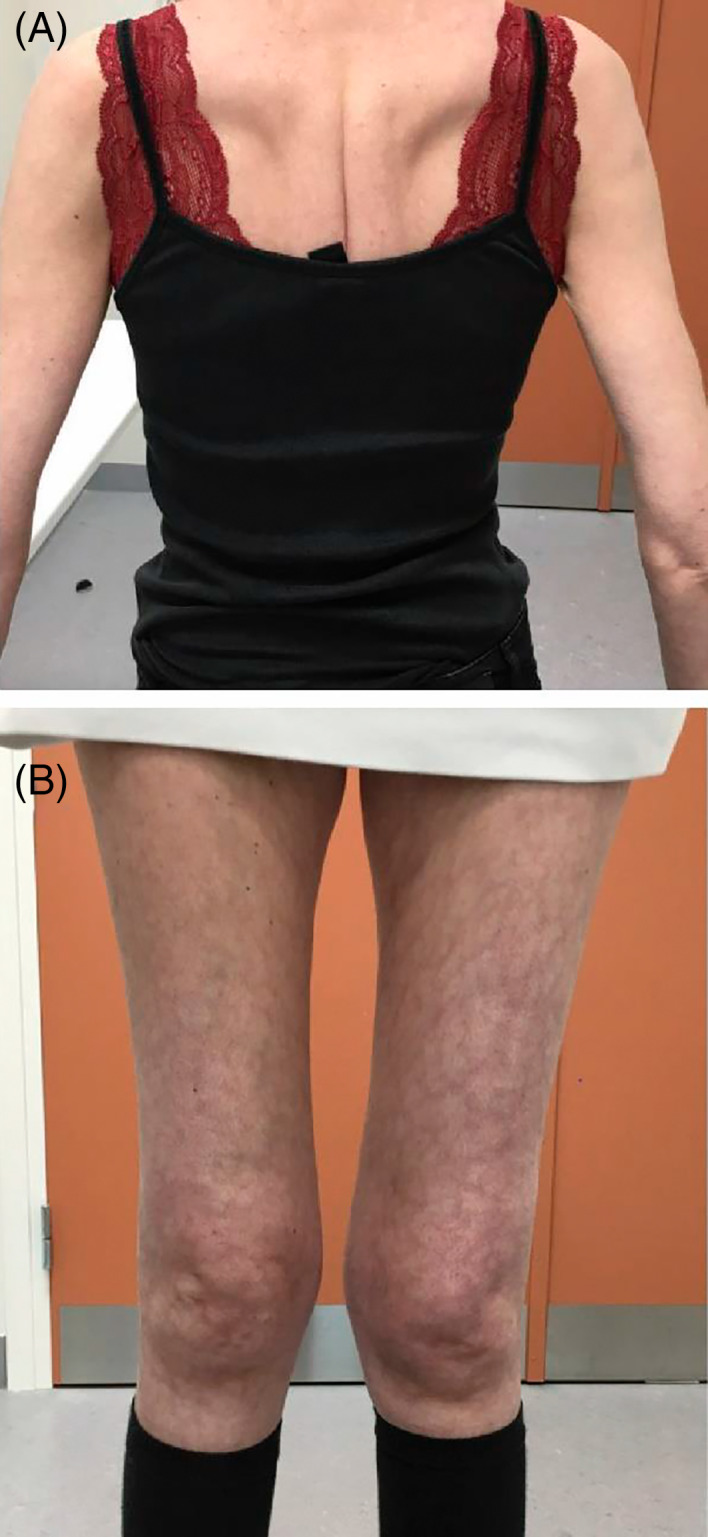
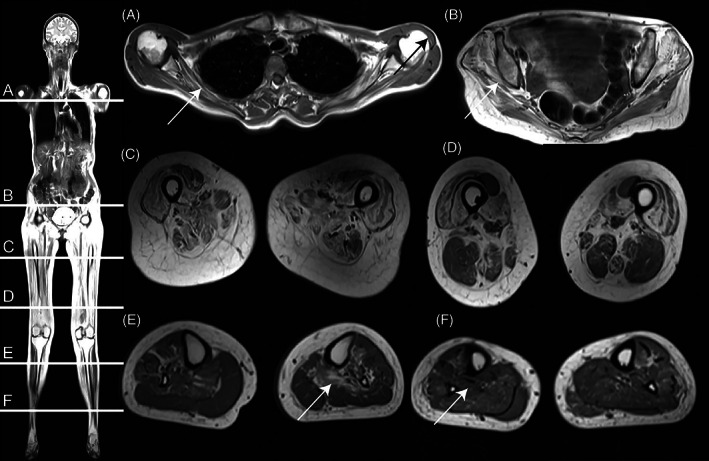
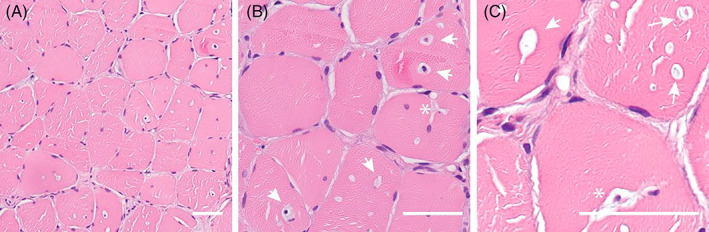
Deficiency of the enzyme β-galactosidase due to variants in the GLB1-gene is associated with metabolic disorders: Morquio B and GM1-gangliosidosis. Here, we report a case compound heterozygous for variants in the GLB1-gene and a severe muscular phenotype. Full body T1-w MRI was conducted for muscular involvement. Biopsy was stained with hematoxylin and eosin for histopathological evaluation. EDTA blood-sample was subjected to whole exome sequencing. Metabolic analysis included residual enzyme activity and evaluation urinary substrate secretion. Additionally, electroneurography, echocardiography, forced volume capacity and biochemistry were evaluated. Examination showed severe proximal weakness (MRC: hip flexion 2, hip extension 2, and shoulder rotation 2), Gower's sign, no extrapyramidal symptoms and normal creatine kinase levels. MRI showed severe muscle wasting of the thigh and shoulder girdle. Muscle biopsy showed mild myopathic changes. β-galactosidase activity was reduced to 28%-34%. Urinary glycosaminoglycan was elevated by 5.9-8.6 mg/mmol (ref.:0-5.1 mg/mmol). Electrophoresis indicated excess keratan sulfate. Exome sequencing revealed two missense variants in the GLB1 gene. Clinical features, genetic testing and laboratory findings indicate a case of β-galactosidase-deficiency with a muscular phenotype.
Keywords: GLB1‐related disorders; Morquio B; fat replacement; metabolic neuromuscular disorder; muscle wasting; myopathy; β‐galactosidase deficiency.
© 2022 The Authors. JIMD Reports published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Jonas Jalili Pedersen, Morten Duno, Flemming Wibrand, Christian Hammer, Thomas Krag, John Vissing declare that they have no conflict of interest.
Figures
References
-
- Regier DS, Tifft CJ, Rothermel CE. GLB1‐Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews® [Internet]. University of Washington; 1993. - PubMed
-
- Shimada T, Tomatsu S, Mason RW, et al. Di‐sulfated Keratan sulfate as a novel biomarker for Mucopolysaccharidosis II, IVA, and IVB. In: Zschocke J, Baumgartner M, Morava E, Patterson M, Rahman S, Peters V, eds. JIMD Reports, Volume 21. Vol 21. Springer; 2014:1‐13. doi: 10.1007/8904_2014_330 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
