

Functional genomics of OCTN2 variants informs protein-specific variant effect predictor for Carnitine Transporter Deficiency
- PMID: 36343260
- PMCID: PMC9674959
- DOI: 10.1073/pnas.2210247119
Functional genomics of OCTN2 variants informs protein-specific variant effect predictor for Carnitine Transporter Deficiency
Abstract
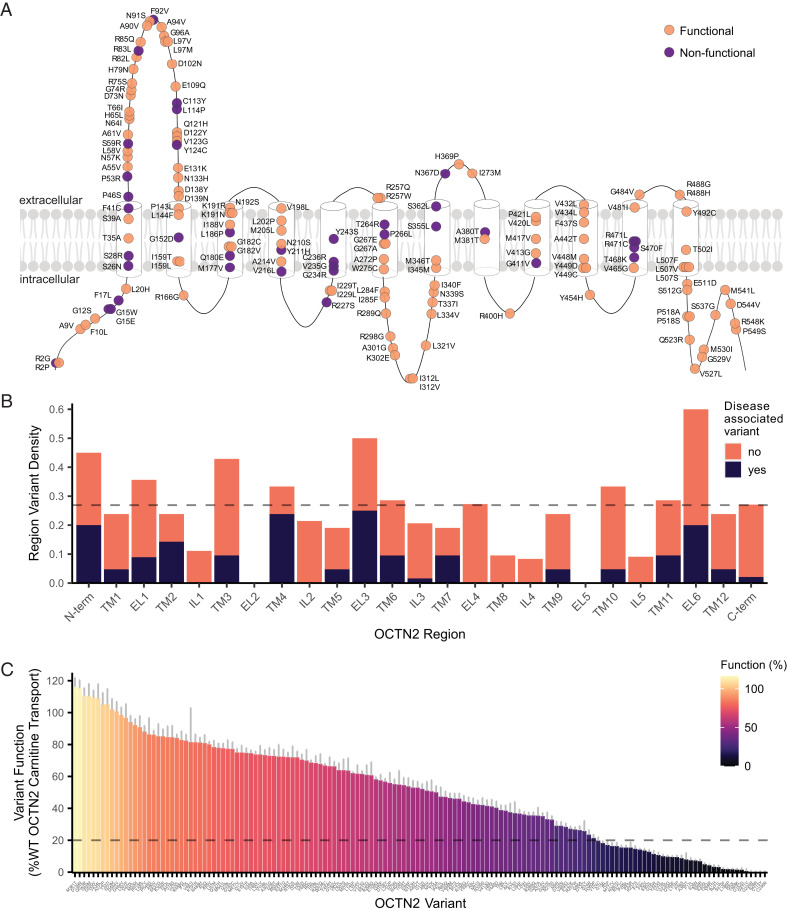
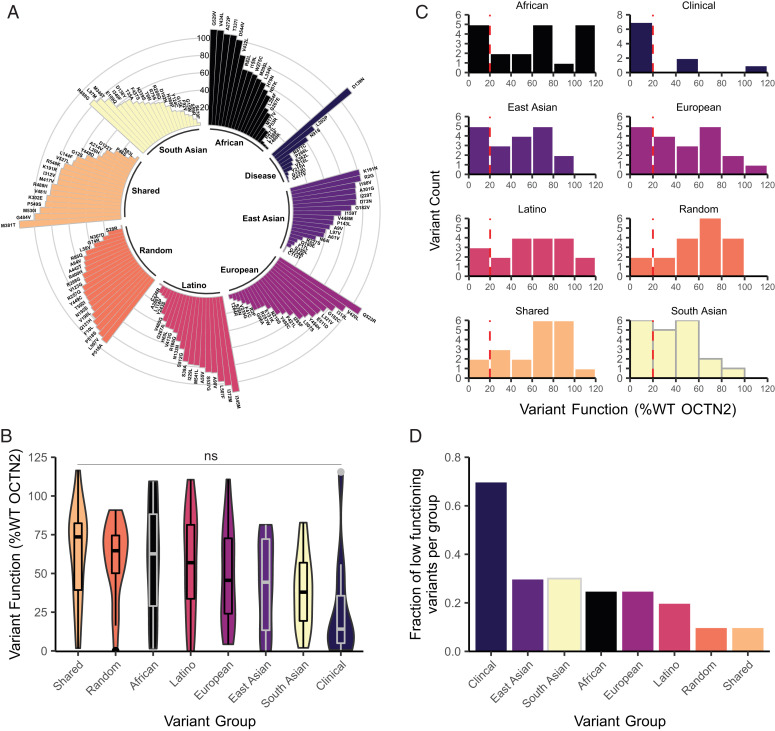
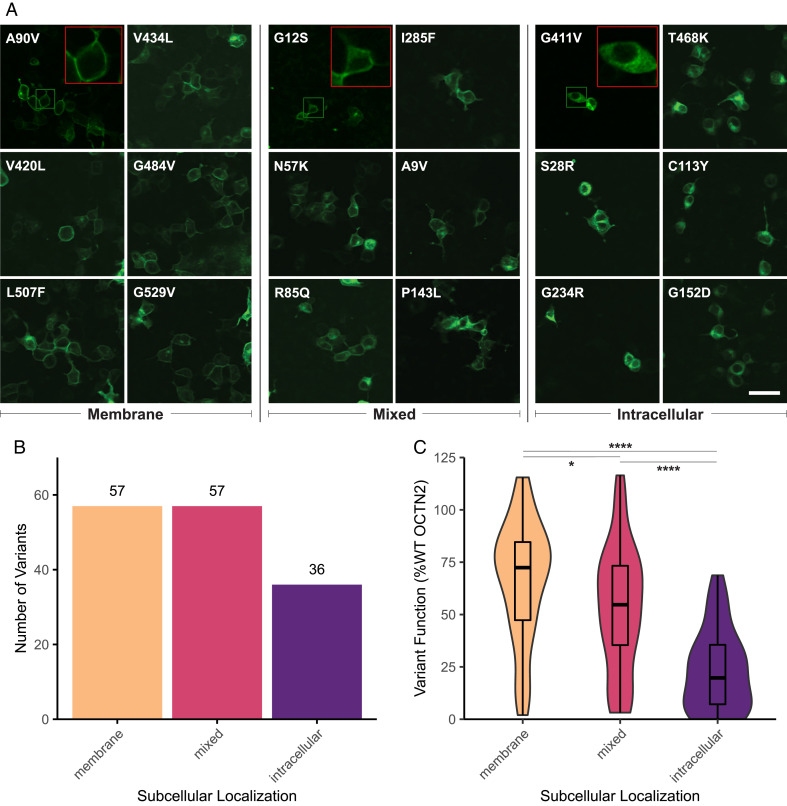
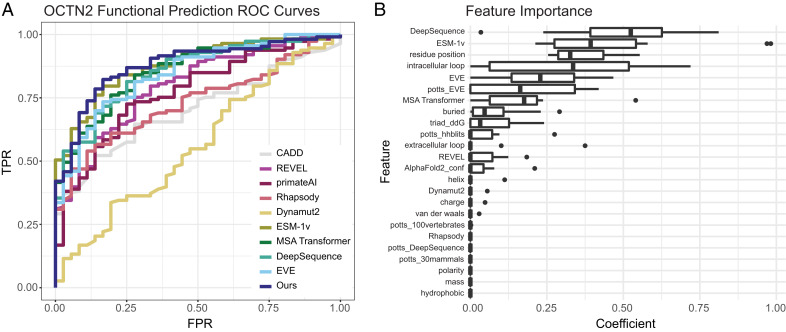
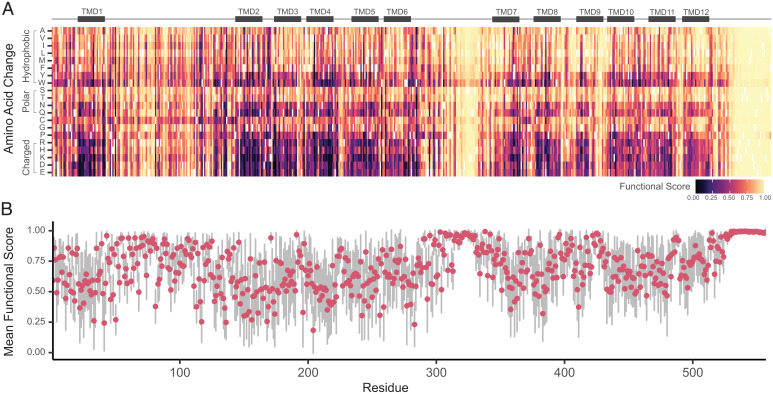
Genetic variants in SLC22A5, encoding the membrane carnitine transporter OCTN2, cause the rare metabolic disorder Carnitine Transporter Deficiency (CTD). CTD is potentially lethal but actionable if detected early, with confirmatory diagnosis involving sequencing of SLC22A5. Interpretation of missense variants of uncertain significance (VUSs) is a major challenge. In this study, we sought to characterize the largest set to date (n = 150) of OCTN2 variants identified in diverse ancestral populations, with the goals of furthering our understanding of the mechanisms leading to OCTN2 loss-of-function (LOF) and creating a protein-specific variant effect prediction model for OCTN2 function. Uptake assays with 14C-carnitine revealed that 105 variants (70%) significantly reduced transport of carnitine compared to wild-type OCTN2, and 37 variants (25%) severely reduced function to less than 20%. All ancestral populations harbored LOF variants; 62% of green fluorescent protein (GFP)-tagged variants impaired OCTN2 localization to the plasma membrane of human embryonic kidney (HEK293T) cells, and subcellular localization significantly associated with function, revealing a major LOF mechanism of interest for CTD. With these data, we trained a model to classify variants as functional (>20% function) or LOF (<20% function). Our model outperformed existing state-of-the-art methods as evaluated by multiple performance metrics, with mean area under the receiver operating characteristic curve (AUROC) of 0.895 ± 0.025. In summary, in this study we generated a rich dataset of OCTN2 variant function and localization, revealed important disease-causing mechanisms, and improved upon machine learning-based prediction of OCTN2 variant function to aid in variant interpretation in the diagnosis and treatment of CTD.
Keywords: machine learning; rare disease; transporter; variant interpretation.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Online Mendelian Inheritance in Man (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD: ).
-
- Rinaldo P., Stanley C. A., Hsu B. Y., Sanchez L. A., Stern H. J., Sudden neonatal death in carnitine transporter deficiency. J. Pediatr. 131, 304–305 (1997). - PubMed
-
- Rasmussen J., et al. , Increased risk of sudden death in untreated primary carnitine deficiency. J. Inherit. Metab. Dis. 43, 290–296 (2020). - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
