

Genotype-Tailored ERK/MAPK Pathway and HDAC Inhibition Rewires the Apoptotic Rheostat to Trigger Colorectal Cancer Cell Death
- PMID: 36343387
- PMCID: PMC9808369
- DOI: 10.1158/1535-7163.MCT-22-0101
Genotype-Tailored ERK/MAPK Pathway and HDAC Inhibition Rewires the Apoptotic Rheostat to Trigger Colorectal Cancer Cell Death
Abstract
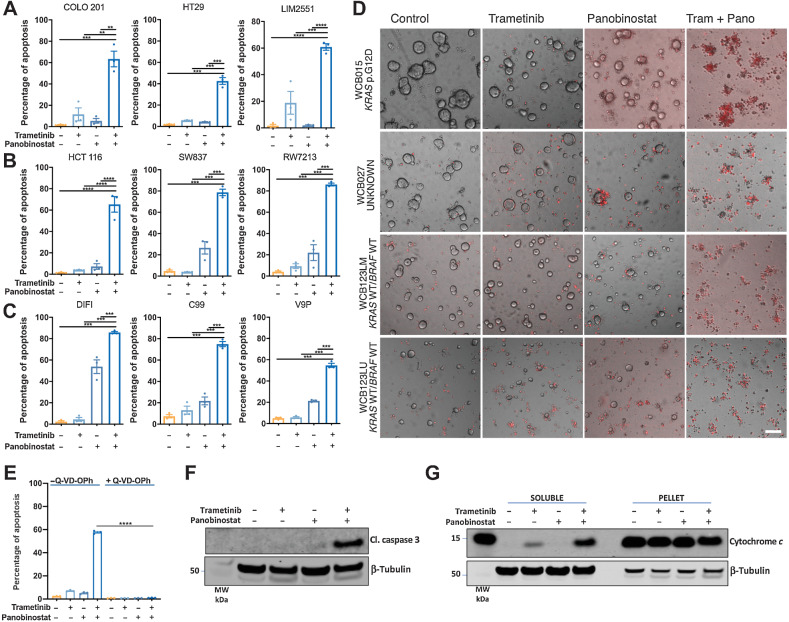
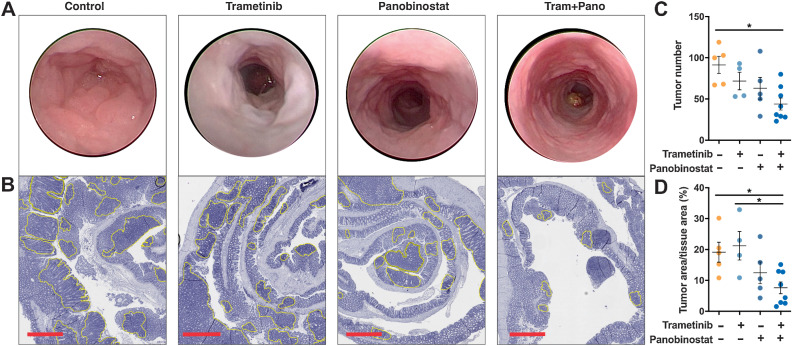
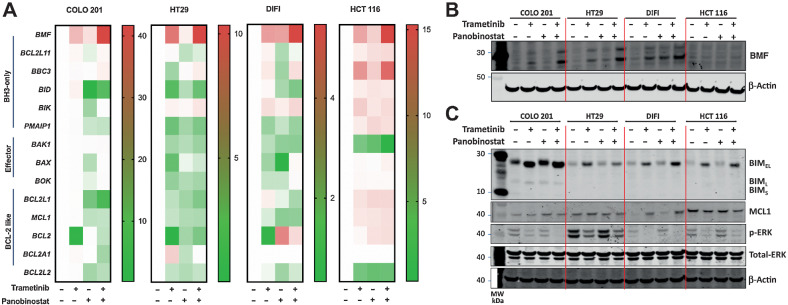
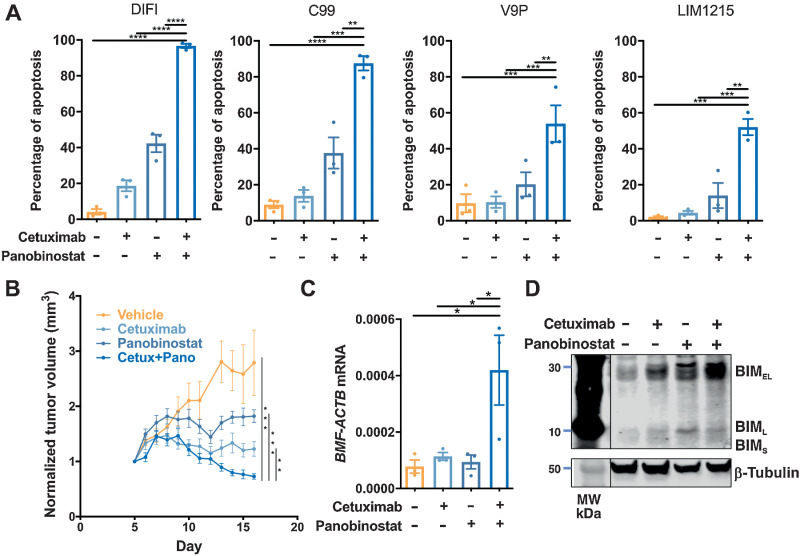
The EGFR/RAS/MEK/ERK signaling pathway (ERK/MAPK) is hyperactivated in most colorectal cancers. A current limitation of inhibitors of this pathway is that they primarily induce cytostatic effects in colorectal cancer cells. Nevertheless, these drugs do induce expression of proapoptotic factors, suggesting they may prime colorectal cancer cells to undergo apoptosis. As histone deacetylase inhibitors (HDACis) induce expression of multiple proapoptotic proteins, we examined whether they could synergize with ERK/MAPK inhibitors to trigger colorectal cancer cell apoptosis. Combined MEK/ERK and HDAC inhibition synergistically induced apoptosis in colorectal cancer cell lines and patient-derived tumor organoids in vitro, and attenuated Apc-initiated adenoma formation in vivo. Mechanistically, combined MAPK/HDAC inhibition enhanced expression of the BH3-only proapoptotic proteins BIM and BMF, and their knockdown significantly attenuated MAPK/HDAC inhibitor-induced apoptosis. Importantly, we demonstrate that the paradigm of combined MAPK/HDAC inhibitor treatment to induce apoptosis can be tailored to specific MAPK genotypes in colorectal cancers, by combining an HDAC inhibitor with either an EGFR, KRASG12C or BRAFV600 inhibitor in KRAS/BRAFWT; KRASG12C, BRAFV600E colorectal cancer cell lines, respectively. These findings identify a series of ERK/MAPK genotype-tailored treatment strategies that can readily undergo clinical testing for the treatment of colorectal cancer.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
![Figure 4. Effect of dual inactivation of BIM and BMF on trametinib and panobinostat induced apoptosis. COLO 201 BIM deletion CRISPR cells [minus doxycycline (−DOX) and plus doxycycline (+DOX)] were transiently transfected with non-targeting or BMF-targeting siRNAs for 24 hours. Cells were then treated with trametinib (10 nmol/L) and panobinostat (25 nmol/L) alone and in combination for 24 hours. A, Knockdown efficiency of BMF as determined by qRT-PCR. B, Validation of CRISPR-Cas9–mediated BIM deletion by Western blot. β-Tubulin was used as a loading control. C, Apoptosis determined by propidium iodide staining and FACS analysis. The values shown are mean ± SEM from a single experiment performed in technical triplicate. Similar results were obtained in a second independent experiment. One-way ANOVA, with Tukey's multiple comparison testing; ****, P ≤ 0.0001)](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/3ad7/9808369/f7467f55271a/52fig4.jpg)
References
-
- McKay JA, Murray LJ, Curran S, Ross VG, Clark C, Murray GI, et al. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur J Cancer 2002;38:2258–64. - PubMed
-
- Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life, and fate. Nat Rev Mol Cell Biol 2020;21:607–32. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
