

Connecting multiple microenvironment proteomes uncovers the biology in head and neck cancer
- PMID: 36344512
- PMCID: PMC9640649
- DOI: 10.1038/s41467-022-34407-1
Connecting multiple microenvironment proteomes uncovers the biology in head and neck cancer
Abstract
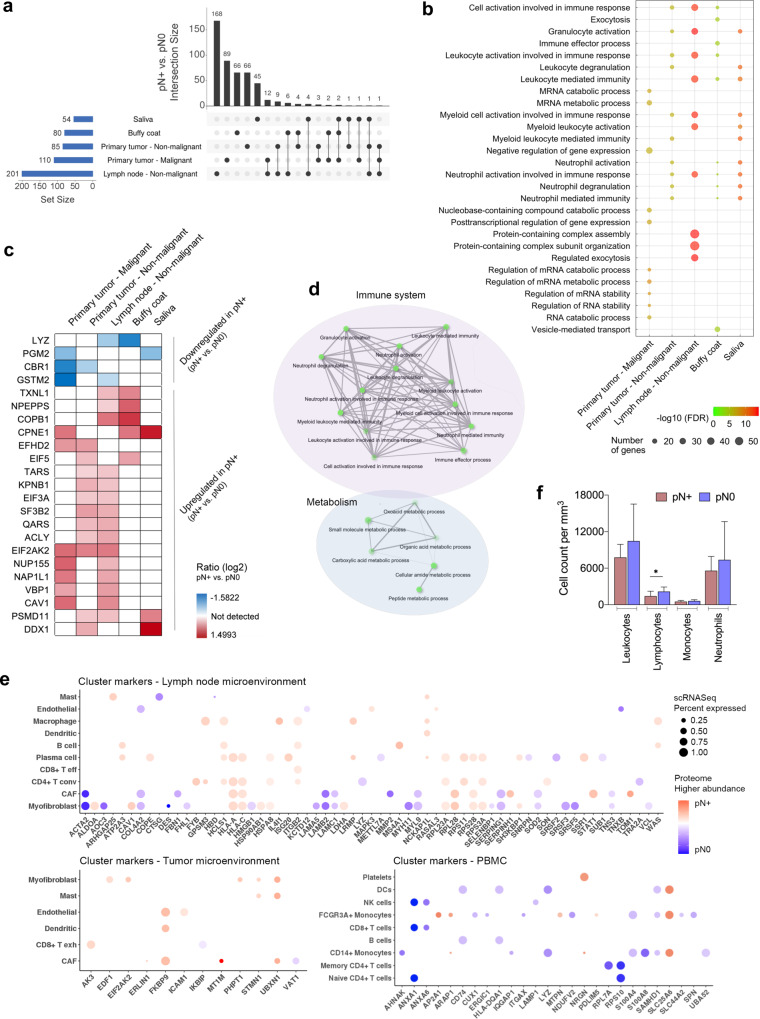
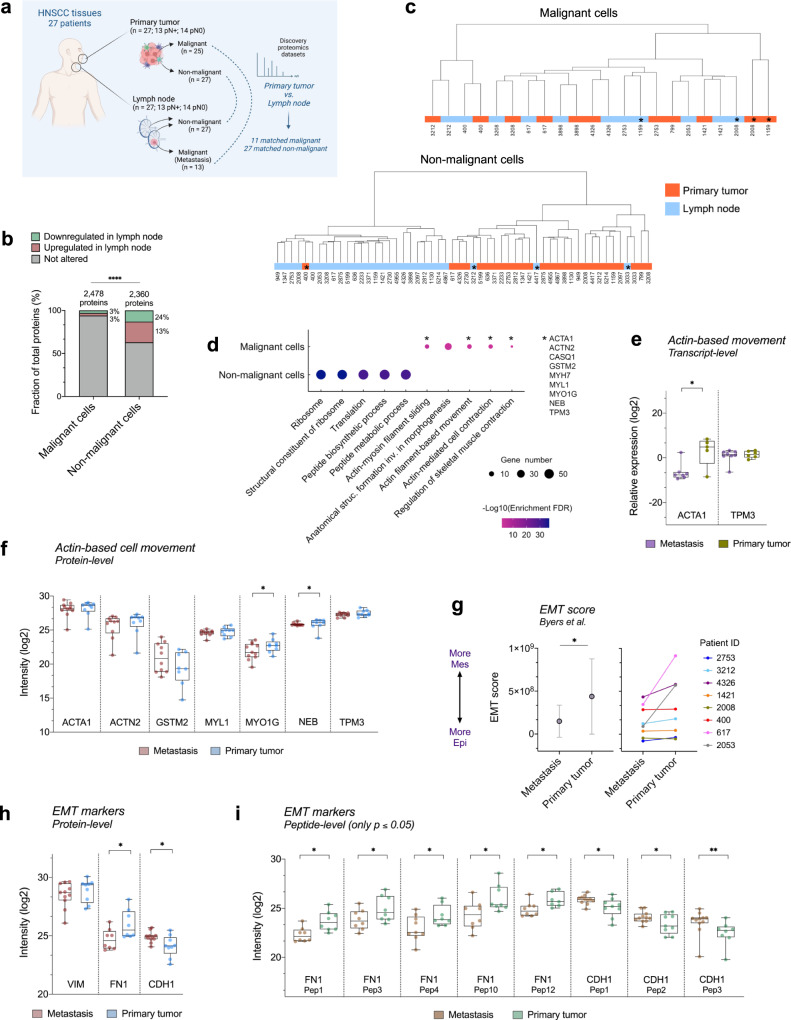
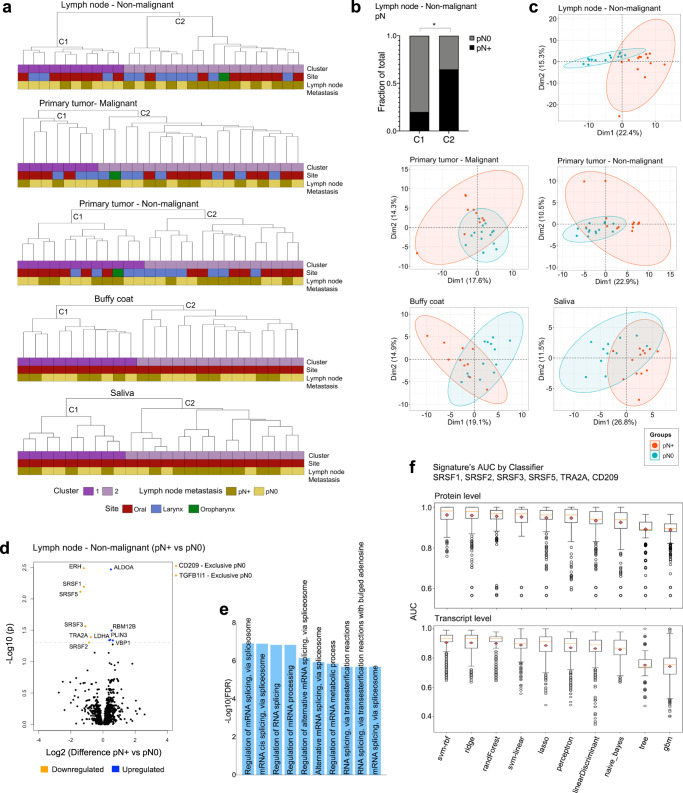
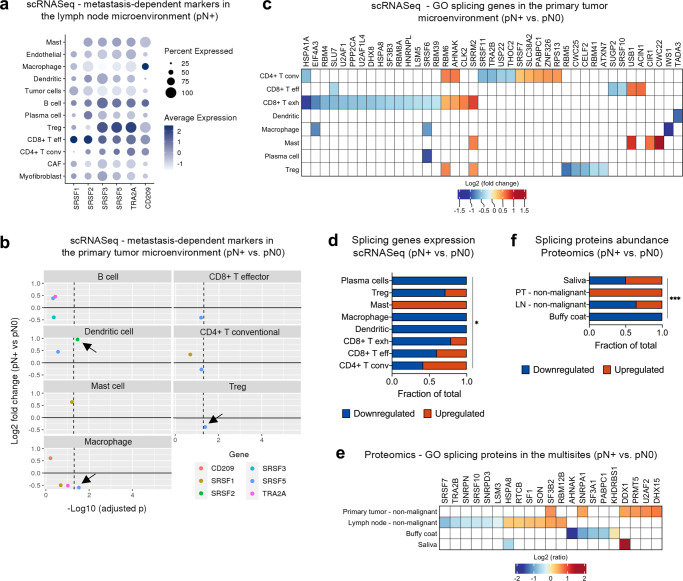
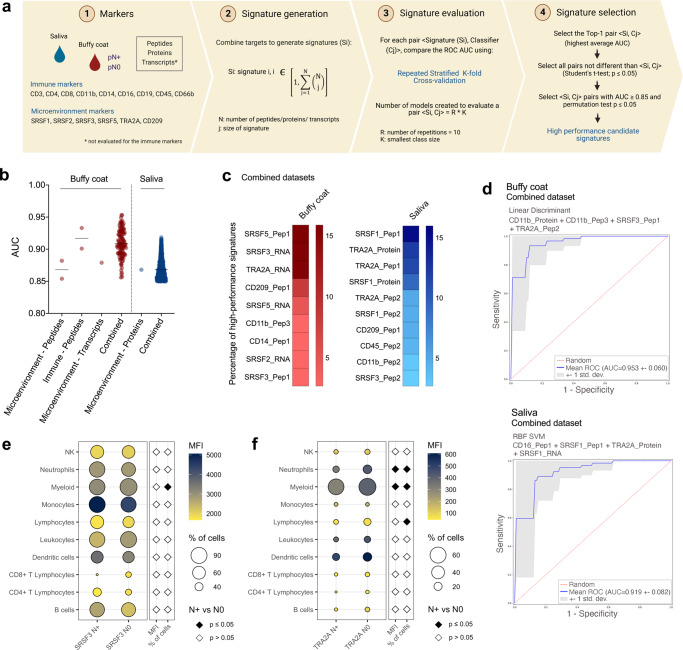
The poor prognosis of head and neck cancer (HNC) is associated with metastasis within the lymph nodes (LNs). Herein, the proteome of 140 multisite samples from a 59-HNC patient cohort, including primary and matched LN-negative or -positive tissues, saliva, and blood cells, reveals insights into the biology and potential metastasis biomarkers that may assist in clinical decision-making. Protein profiles are strictly associated with immune modulation across datasets, and this provides the basis for investigating immune markers associated with metastasis. The proteome of LN metastatic cells recapitulates the proteome of the primary tumor sites. Conversely, the LN microenvironment proteome highlights the candidate prognostic markers. By integrating prioritized peptide, protein, and transcript levels with machine learning models, we identify nodal metastasis signatures in blood and saliva. We present a proteomic characterization wiring multiple sites in HNC, thus providing a promising basis for understanding tumoral biology and identifying metastasis-associated signatures.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
