

An organ-on-chip model of pulmonary arterial hypertension identifies a BMPR2-SOX17-prostacyclin signalling axis
- PMID: 36344664
- PMCID: PMC9640600
- DOI: 10.1038/s42003-022-04169-z
An organ-on-chip model of pulmonary arterial hypertension identifies a BMPR2-SOX17-prostacyclin signalling axis
Abstract
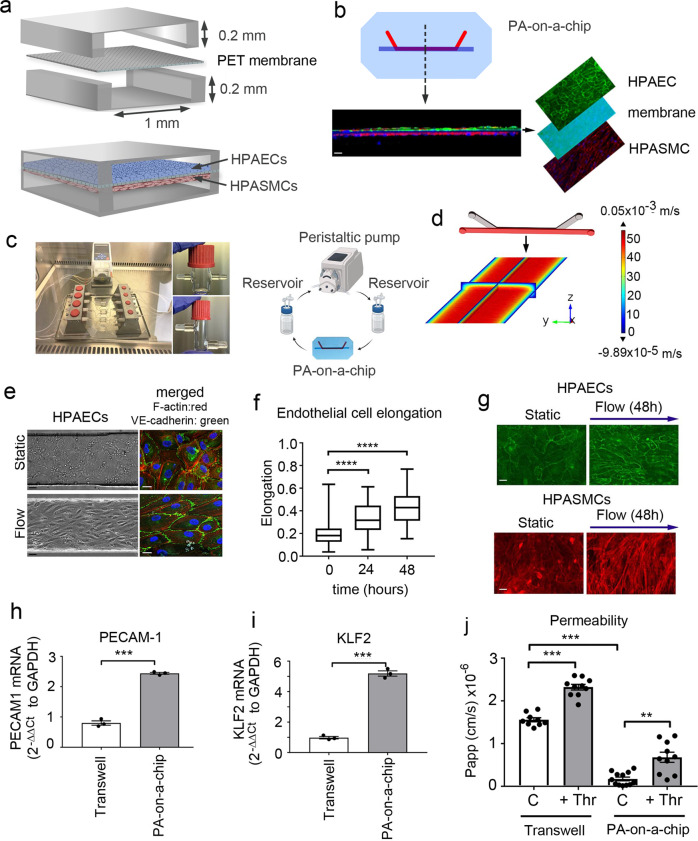
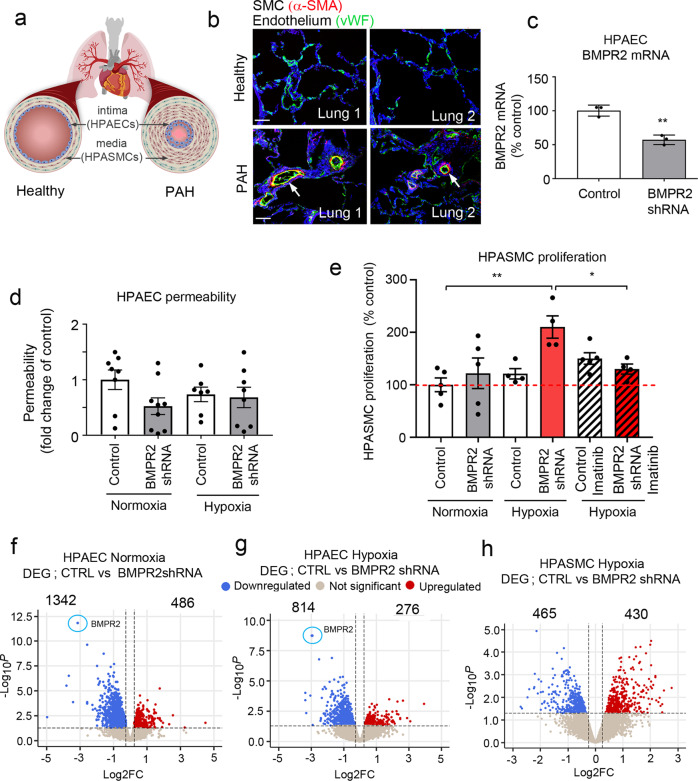
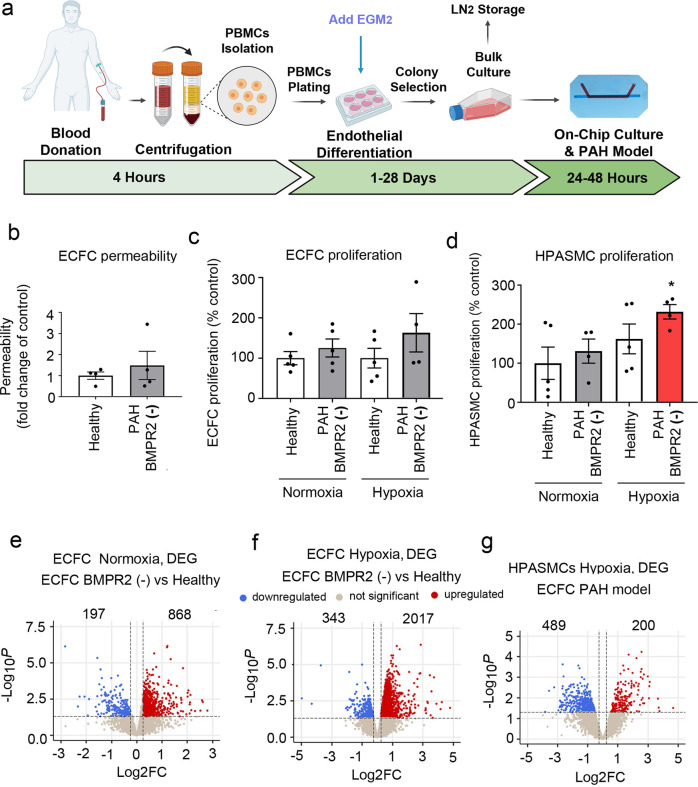
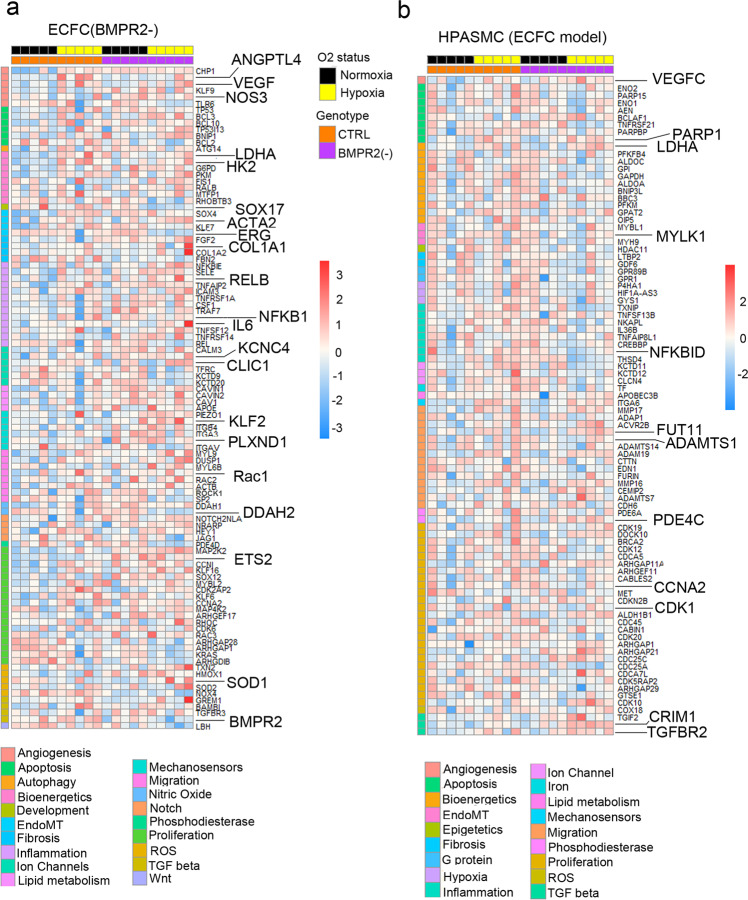
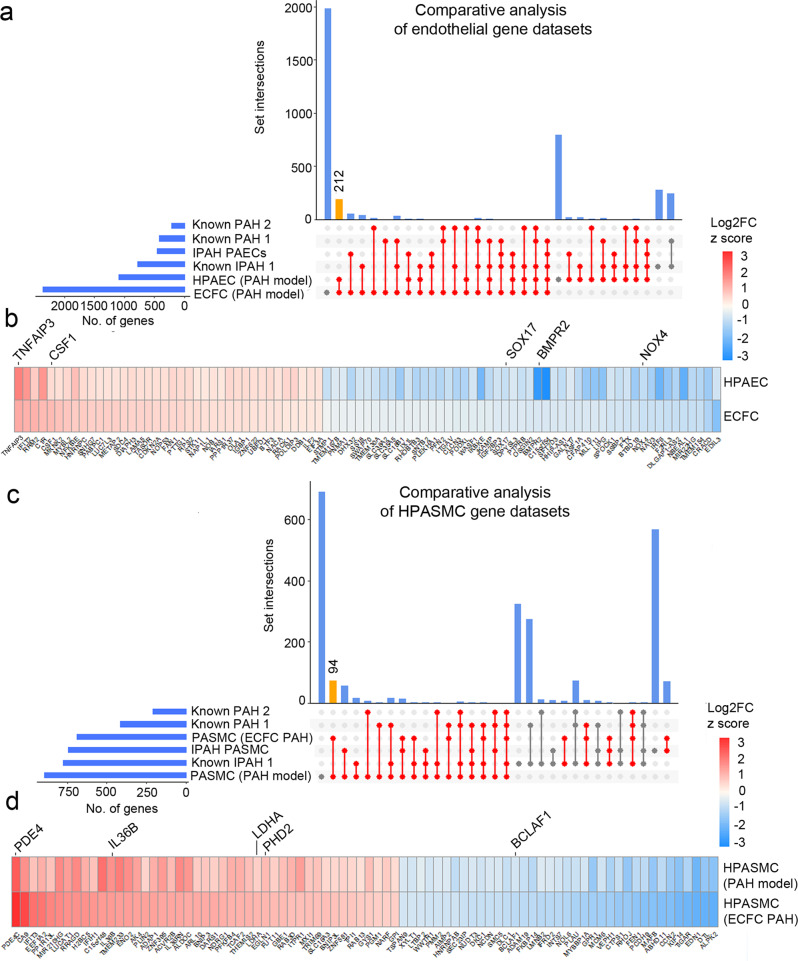
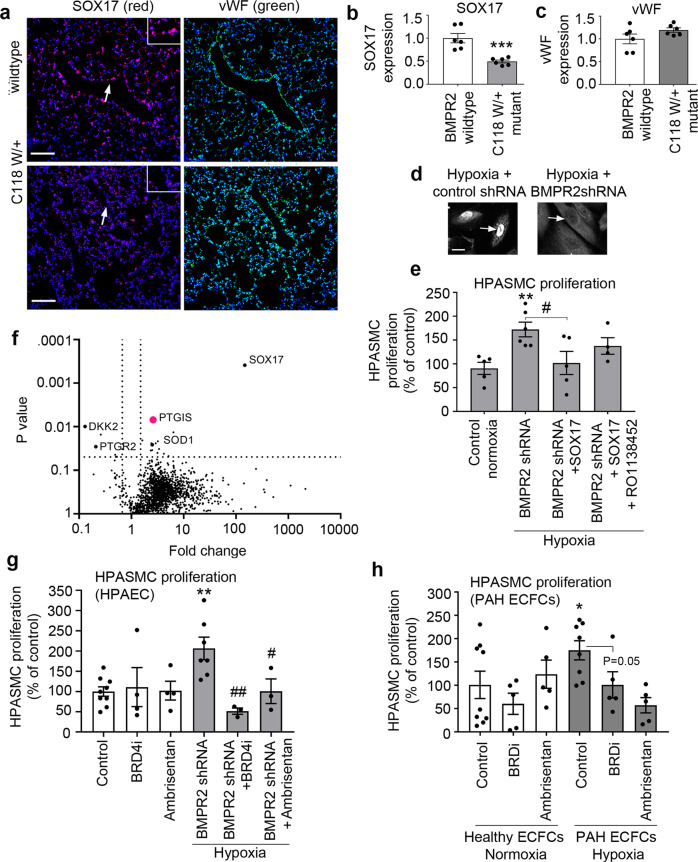
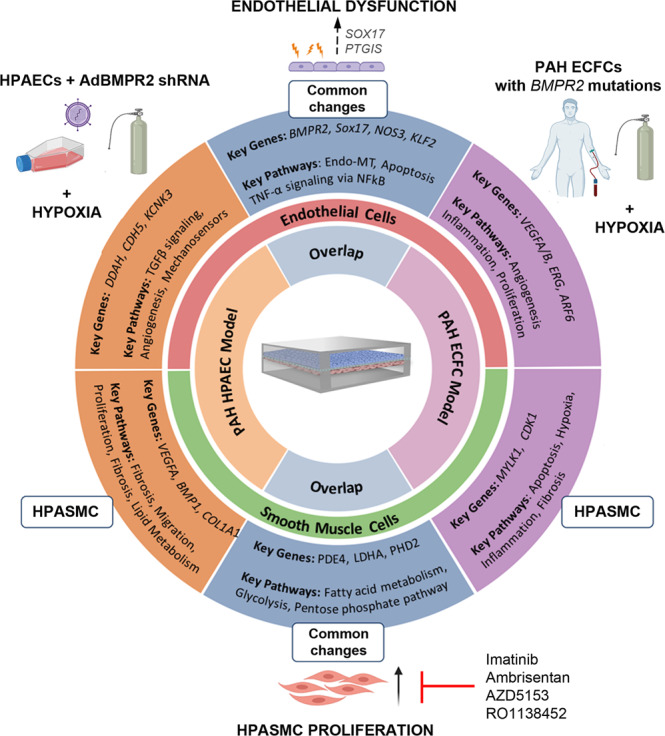
Pulmonary arterial hypertension (PAH) is an unmet clinical need. The lack of models of human disease is a key obstacle to drug development. We present a biomimetic model of pulmonary arterial endothelial-smooth muscle cell interactions in PAH, combining natural and induced bone morphogenetic protein receptor 2 (BMPR2) dysfunction with hypoxia to induce smooth muscle activation and proliferation, which is responsive to drug treatment. BMPR2- and oxygenation-specific changes in endothelial and smooth muscle gene expression, consistent with observations made in genomic and biochemical studies of PAH, enable insights into underlying disease pathways and mechanisms of drug response. The model captures key changes in the pulmonary endothelial phenotype that are essential for the induction of SMC remodelling, including a BMPR2-SOX17-prostacyclin signalling axis and offers an easily accessible approach for researchers to study pulmonary vascular remodelling and advance drug development in PAH.
© 2022. The Author(s).
Conflict of interest statement
Ambrisentan and AZD5153 used in this study were provided by Astra Zeneca.
Figures

References
-
- Hoeper MM, et al. A global view of pulmonary hypertension. Lancet Respir. Med. 2016;4:306–322. - PubMed
-
- Yuan JX, Rubin LJ. Pathogenesis of pulmonary arterial hypertension: the need for multiple hits. Circulation. 2005;111:534–538. - PubMed
-
- Southgate L, Machado RD, Graf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2020;17:85–95. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
