

A structural biology community assessment of AlphaFold2 applications
- PMID: 36344848
- PMCID: PMC9663297
- DOI: 10.1038/s41594-022-00849-w
A structural biology community assessment of AlphaFold2 applications
Abstract
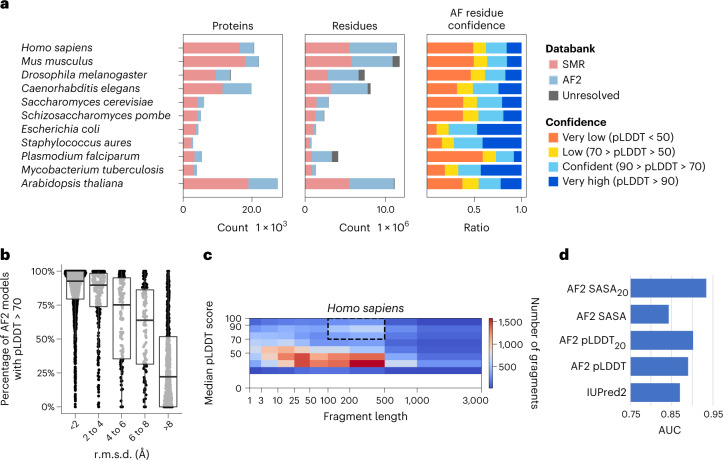
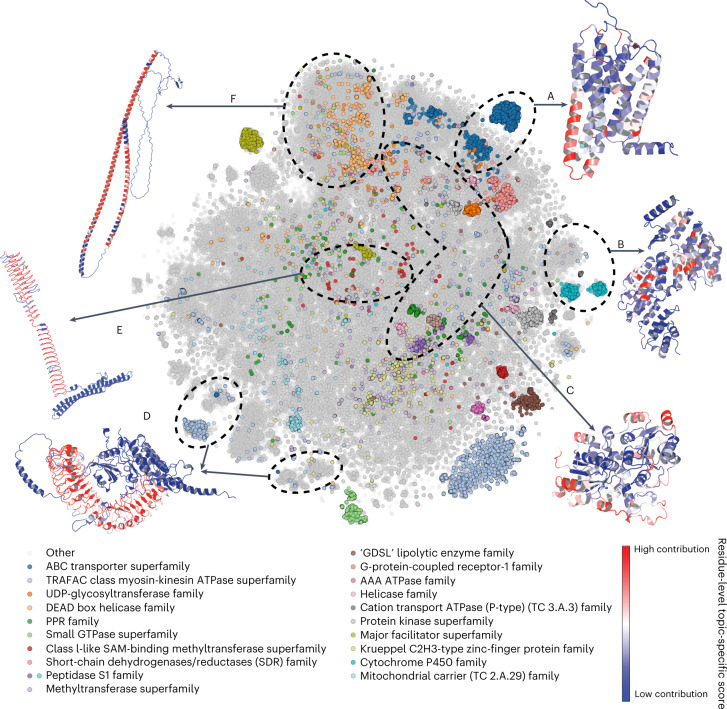
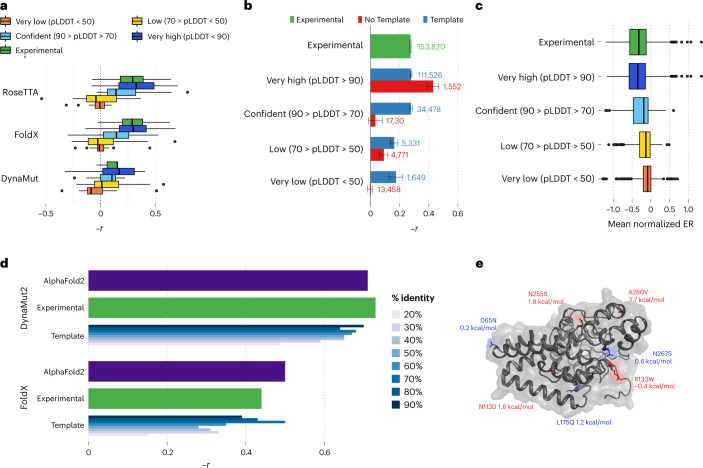
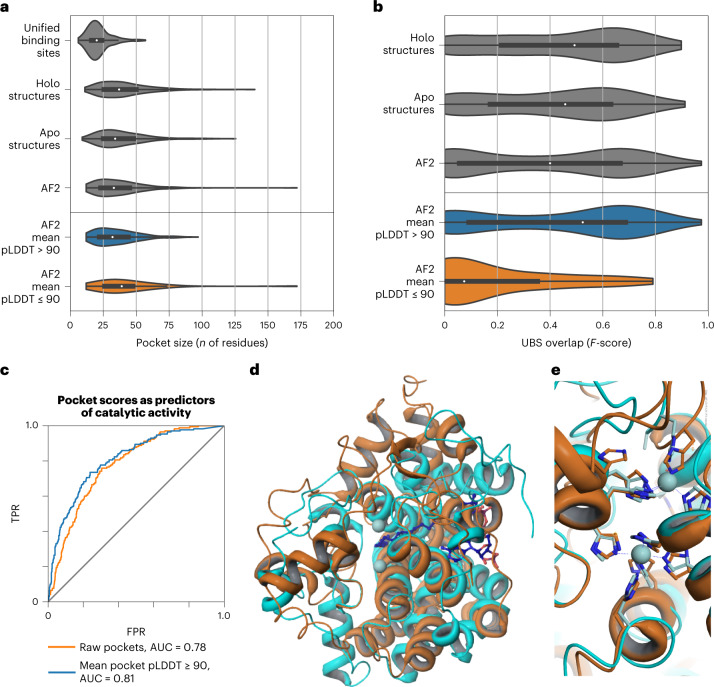
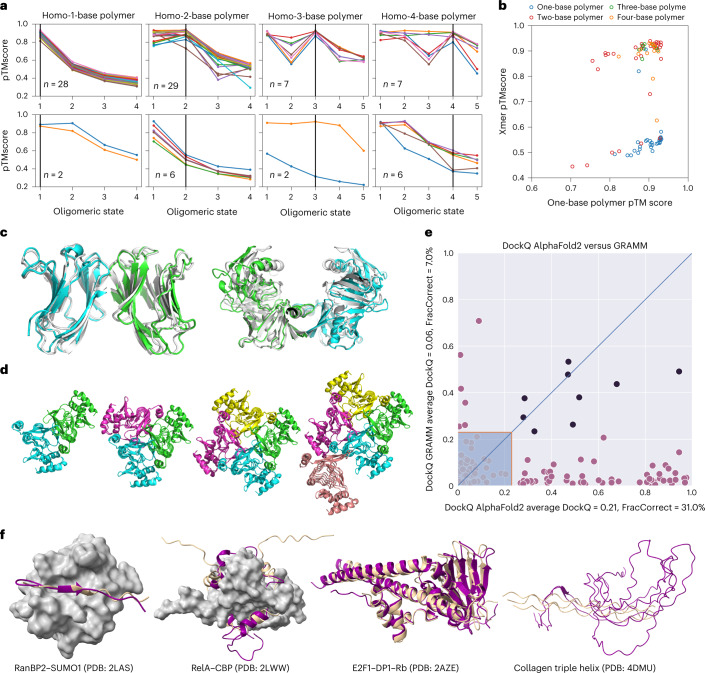
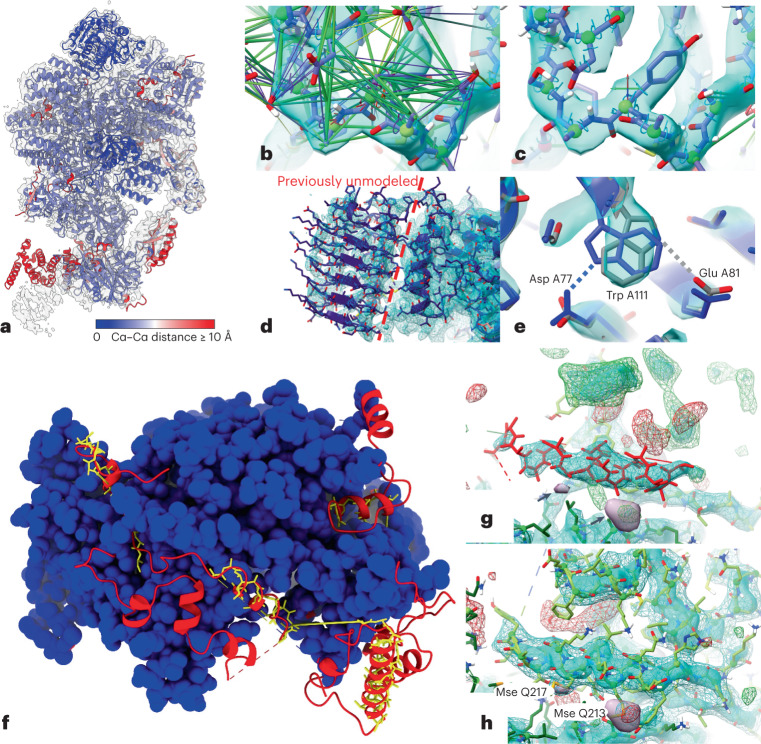
Most proteins fold into 3D structures that determine how they function and orchestrate the biological processes of the cell. Recent developments in computational methods for protein structure predictions have reached the accuracy of experimentally determined models. Although this has been independently verified, the implementation of these methods across structural-biology applications remains to be tested. Here, we evaluate the use of AlphaFold2 (AF2) predictions in the study of characteristic structural elements; the impact of missense variants; function and ligand binding site predictions; modeling of interactions; and modeling of experimental structural data. For 11 proteomes, an average of 25% additional residues can be confidently modeled when compared with homology modeling, identifying structural features rarely seen in the Protein Data Bank. AF2-based predictions of protein disorder and complexes surpass dedicated tools, and AF2 models can be used across diverse applications equally well compared with experimentally determined structures, when the confidence metrics are critically considered. In summary, we find that these advances are likely to have a transformative impact in structural biology and broader life-science research.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Burley SK, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49:D437–D451. - PMC - PubMed
-
- Thomas J, Ramakrishnan N, Bailey-Kellogg C. Graphical models of residue coupling in protein families. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008;5:183–197. - PubMed
-
- Dunn SD, Wahl LM, Gloor GB. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. - PubMed
-
- Bartlett GJ, Taylor WR. Using scores derived from statistical coupling analysis to distinguish correct and incorrect folds in de-novo protein structure prediction. Proteins. 2008;71:950–959. - PubMed
